БИОХИМИЯ УЧЕБНИК ДЛЯ ВУЗОВ - Е. С. Северина - 2004
РАЗДЕЛ 9. ОБМЕН И ФУНКЦИИ АМИНОКИСЛОТ
VIII. Обмен отдельных аминокислот
Кроме общих путей обмена, характерных для большинства аминокислот, существуют и специфические пути превращения почти всех аминокислот, входящих в состав белков. Рассмотрим обмен некоторых аминокислот, пути превращения которых приводят к синтезу биологически активных продуктов и во многом определяют физиологические состояния в организме человека.
А. Обмен серина и глицина
Серин — заменимая аминокислота, синтезируется из промежуточного продукта гликолиза — 3-фосфоглицерата, а аминогруппу получает от глутаминовой кислоты.
Глицин — также заменимая аминокислота, основным источником которой служит серин. Реакцию синтеза глицина из серина катализирует фермент серин-оксиметилтрансфераза, коферментом которой является Н4-фолат (см. схему А).

Реакция превращения серина в глицин легко обратима. Основной путь катаболизма глицина у человека и других позвоночных также связан с использованием Н4-фолата (см. схему Б).
Эта реакция обратима и катализируется глицинсинтазой — ферментным комплексом, похожим на пируватдегидрогеназный комплекс, и локализованным в митохондриях клеток печени. По последним данным глицинрасщепляющая ферментная система несколько отличается от глицинсинтазы и содержит 4 белка: Р-белок, включающий кофермент ПФ, Н-белок, содержащий липоевую кислоту, Т-белок с коферментом Н4-фолат, L-белок, являющийся дигидролипоилдегидрогеназой с коферментом NАD+.
1. Пути метаболизма серина и глицина
Аминокислоты серин и глицин выполняют в организме человека разнообразные и очень важные функции. Роль серина и глицина в синтезе многих биологически важных соединений представлена на рис. 9-24.
Рис. 9-24. Биологическая роль серина и глицина.

На рисунке видно, что обе аминокислоты необходимы не только для синтеза белков и глюкозы (при её недостатке в клетках), но и нуклеотидов, коферментов, гема, сложных липидов, креатина и других соединений. Многие из этих реакций представлены в соответствующих разделах учебника.
2. Роль фолиевой кислоты в обмене аминокислот
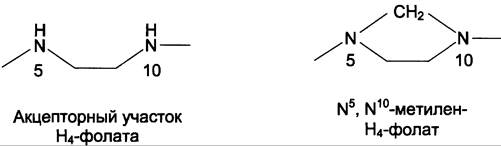
В превращениях серина и глицина главную роль играют ферменты, коферментами которых служат производные фолиевой кислоты. Этот витамин широко распространён в животных и растительных пищевых продуктах (см. раздел 3). Молекула фолиевой кислоты (фолата) состоит из 3 частей: птеринового производного, парааминобензойной и глутаминовой кислот (см. схему А).

Фолиевую кислоту (фолат) называют также птероилглутаминовой кислотой. Птерины широко распространены в природе. Некоторые из них, например, ксантоптерин, являются пигментами глаз и крыльев насекомых (бабочек).
Коферментную функцию выполняет восстановленная форма фолата — тетрагидрофолиевая кислота (ТГФК или Н4-фолат) (см. схему Б).

Фолиевая кислота в печени превращается в Н4-фолат в несколько стадий с участием ферментов фолатредуктазы и дигидрофолатредуктазы, коферментом которых служит NADPH.
Н4-фолат — акцептор β-углеродного атома серина. При этом образуется метиленовый мостик между атомами азота в молекуле Н4-фолата в положениях 5 и 10, образуя метилен-Н4-фолат (см. схему В).
3. Образование и использование одноуглеродных фрагментов
Особое значение реакций катаболизма серина и глицина заключается в том, что они сопровождаются образованием одноуглеродного метиленового фрагмента (-СН2-). Метиленовая группа в молекуле метилен-Н4-фолата может превращаться в другие одноуглеродные группы (фрагменты): метенильную (-СН=), формильную (-НС=О), метальную (-СН3) и формими- ногруппу (-СН=NН) (рис. 9-25).
Рис. 9-25. Образование производных Н4-фолата.
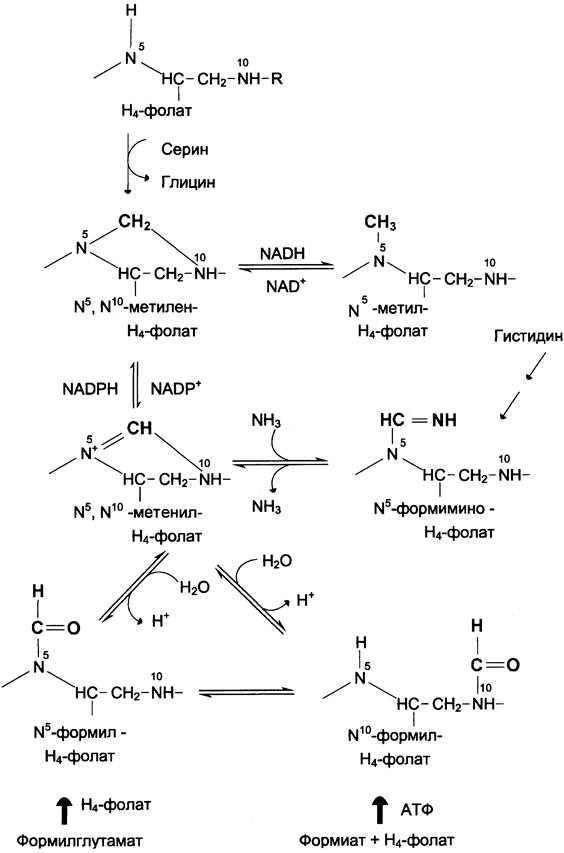
Ещё один источник формильного и формимино-фрагментов — гистидин. Катаболизм гистидина происходит только в печени (очень небольшой процент в коже) в результате следующих реакций (см. схему).

Конечными продуктами катаболизма гистидина являются глутамат, NН3 и одноуглеродные фрагменты — формимино-Н4-фолат и формил-Н4-фолат.
Все образующиеся производные Н4-фолата играют роль промежуточных переносчиков и служат донорами одноуглеродных фрагментов при синтезе некоторых соединений: пуриновых оснований и тимидиловой кислоты (необходимых для синтеза ДНК и РНК), регенерации метионина, синтезе различных формиминопроизводных (формиминоглицина и т. д.) (рис. 9-26).
Рис. 9-26. Образование и использование производных Н4-фолата.

Перенос одноуглеродных фрагментов к акцептору необходим не только для синтеза ряда соединений, но и для регенерации свободного Н4-фолата в печени.
4. Недостаточность фолиевой кислоты
Недостаточность фолиевой кислоты у человека возникает редко (см. раздел 3). Гиповитаминоз фолиевой кислоты приводит к нарушению обмена одноуглеродных фрагментов. Такое же нарушение наблюдается и при недостаточности витамина В12, использование которого связано с обменом фолиевой кислоты (см. подраздел IX).
Первое проявление дефицита фолиевой кислоты — мегалобластная (макроцитарная) анемия. Она характеризуется уменьшением количества эритроцитов, снижением содержания в них гемоглобина, что вызывает увеличение размера эритроцитов. Причина этих симптомов — нарушение синтеза ДНК и РНК из-за недостатка их предшественников — тимидиловой кислоты и пуриновых нуклеотидов вследствие дефицита производных Н4-фолата. Клетки кроветворной ткани быстро делятся, поэтому они в первую очередь реагируют на нарушение синтеза нуклеиновых кислот снижением скорости эритропоэза.
Мегалобластная анемия возникает чаще всего в результате недостаточности фолиевой кислоты и/или витамина В12.
5. Механизм антибактериального действия сульфаниламидных препаратов
Фолиевая кислота является витамином для человека и животных. Однако многие патогенные бактерии способны синтезировать это соединение, используя парааминобензойную кислоту (ПАБК) — одну из составных частей фолата. ПАБК поступает в бактериальные клетки из внешней среды. Сульфаниламидные лекарственные препараты — производные сульфаниламида (белого стрептоцида), похожи по строению на парааминобензойную кислоту. Отличаются они только радикалами (см. схему).
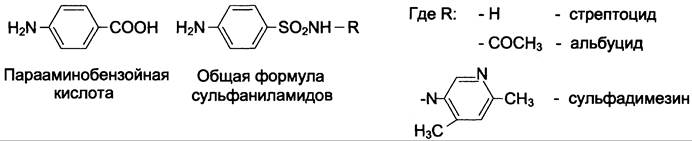
Эти препараты подавляют синтез фолиевой кислоты у бактерий, потому что:
✵ конкурентно ингибируют бактериальные ферменты синтеза фолата, так как являются структурными аналогами парааминобензойной кислоты — одного из субстратов процесса;
✵ могут использоваться как псевдосубстраты из-за относительной субстратной специфичности ферментов, в результате чего синтезируется соединение, похожее на фолиевую кислоту, но не выполняющее её функции.
В обоих случаях в клетках бактерий нарушается обмен одноуглеродных фрагментов и, следовательно, синтез нуклеиновых кислот, что вызывает прекращение размножения бактерий.
В клетках больного сульфаниламидные лекарственные вещества не вызывают подобных изменений, поскольку человек получает с пищей готовую фолиевую кислоту.
6. Наследственные нарушения обмена глицина
В настоящее время известно несколько заболеваний, связанных с нарушениями обмена глицина. В их основе лежит недостаточность какого-либо фермента или дефект системы транспорта этой аминокислоты. Некоторые из этих нарушений представлены ниже.
Гиперглицинемия характеризуется повышенной концентрацией глицина в крови вследствие дефекта глицинрасщепляющей ферментной системы. Наиболее тяжёлое проявление гиперглицинемии — резкое повреждение мозга, судороги, гипотония, нарушение дыхания.
Глицинурия характеризуется повышенным выделением глицина с мочой (до 1 г/сут) при нормальном содержании его в крови. Один из симптомов этого заболевания — образование оксалатных камней в почках, причём содержание оксалата в моче находится в пределах нормы. Избыток оксалата имеет эндогенное происхождение. Скорее всего, он получается из глицина, при дезаминировании которого образуется глиоксилат — предшественник оксалата. Метаболический дефект, очевидно, состоит в нарушении метаболизма глиоксилата — невозможности его превращения снова в глицин из-за дефекта глицинаминотрансферазы. Причиной глицинурии является, очевидно, нарушение реабсорбции глицина в почках. Наследуется как доминантный признак, сцепленный, вероятно, с Х-хромосомой.
Первичная гипероксалатурия характеризуется постоянно высоким выделением оксалата с мочой, независимо от поступления его с пищей. В дальнейшем прогрессирует двустороннее образование оксалатных камней в мочевыводящих путях, развиваются нефрокальциноз и инфекция мочевыводящих путей. Больные погибают в детском возрасте от почечной недостаточности или гипертонии.
Б. Обмен серосодержащих аминокислот
В состав белков человека входят 2 аминокислоты, содержащие серу, — метионин и цистеин. Эти аминокислоты метаболически тесно связаны между собой.
1. Особенности обмена метионина
Метионин — незаменимая аминокислота. Она необходима для синтеза белков организма, участвует в реакциях дезаминирования, является источником атома серы для синтеза цистеина. Метионил-тРНК участвует в инициации процесса трансляции.
Метальная группа метионина — мобильный одноуглеродный фрагмент, используемый для синтеза ряда соединений. Перенос метальной группы метионина на соответствующий акцептор называют реакцией трансметилирования, имеющей важное метаболическое значение.
Метальная группа в молекуле метионина прочно связана с атомом серы, поэтому непосредственным донором этого одноуглеродного фрагмента служит активная форма аминокислоты.
Реакция активации метионина
Активной формой метионина является S-аденозилметионин (SAM) — сульфониевая форма аминокислоты, образующаяся в результате присоединения метионина к молекуле аденозина. Аденозин образуется при гидролизе АТФ (см. схему А).

Эту реакцию катализирует фермент метионин аденозилтрансфераза, присутствующий во всех типах клеток. Структура (-S+-CH3) в SAM — нестабильная группировка, определяющая высокую активность метильной группы (отсюда термин «активный метионин»). Эта реакция уникальна для биологических систем, так как, по-видимому, является единственной известной реакцией, в результате которой освобождаются все три фосфатных остатка АТФ.
Отщепление метильной группы от SAM и перенос её на соединение-акцептор катализируют ферменты метилтрансферазы. SAM в ходе реакции превращается в S-аденозилгомоцистеин (SAГ).
Примеры реакций трансметилирования
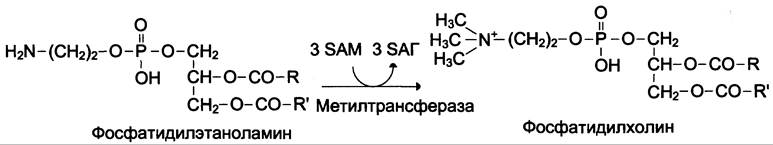
Синтез фосфатидилхолина из фосфатидилэтиноламина
Фосфатидилхолины (лецитины) — наиболее распространённая группа глицерофосфолипидов, участвующих в образовании мембран клеток и липопротеинов, в составе которых осуществляется транспорт липидов (см. раздел 8) (см. схему Б).
Синтез карнитина
Карнитин — переносчик жирных кислот через мембрану митохондрий (см. раздел 8) (см. схему А).
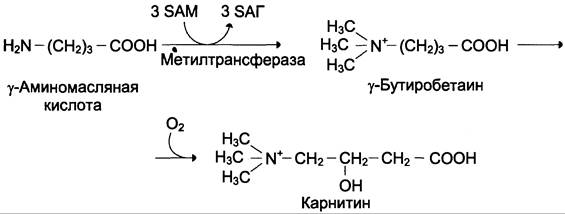
Синтез креатина
Креатин необходим для образования в мышцах высокоэнергетического соединения — креатинфосфата. Синтез креатина идёт в 2 стадии с участием 3 аминокислот: аргинина, глицина и метионина. В почках образуется гуанидин- ацетат при действии глицинамидинотрансферазы (см. схему Б).
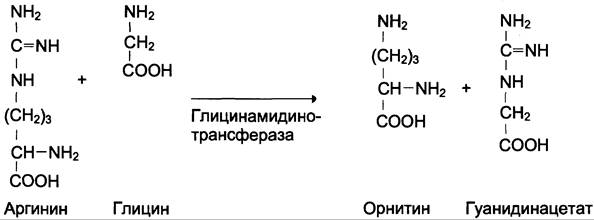
Затем гуанидинацетат транспортируется в печень, где происходит реакция его метилирования (см. схему В).

Креатин с кровотоком переносится в мышцы и клетки мозга, где из него образуется высокоэнергетическое соединение — креатинфосфат. (см. схему А).

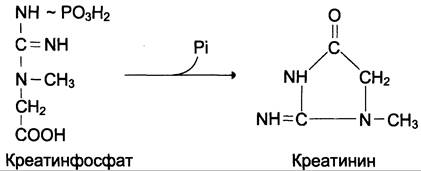
Эта реакция легко обратима и катализируется ферментом креатинкиназой. Фермент локализован в цитозоле и митохондриях клеток, обладает органоспецифичностью. В норме активность его в крови очень мала. Обнаружено три изоферментные формы креатинкиназы (см. раздел 2).
Креатинфосфат играет важную роль в обеспечении энергией работающей мышцы в начальный период. В результате неферментативного дефосфорилирования, главным образом в мышцах, креатинфосфат превращается в креатинин, выводимый с мочой. Суточное выделение креатинина у каждого индивидуума постоянно и пропорционально общей мышечной массе (см. схему Б).
Определение содержания креатина и креатинина в крови и моче используется для характеристики интенсивности работы мышц в спортивной медицине и при некоторых патологических состояниях. Определение активности фермента креатинкиназы и его изоферментных форм в крови используется в медицине для диагностики таких заболеваний, как инфаркт миокарда, миопатии, мышечные дистрофии и др.
Реакции трансметилирования используются так же для:
✵ синтеза адреналина из норадреналина;
✵ синтеза анзерина из карнозина;
✵ метилирования азотистых оснований в нуклеотидах и др. (см. раздел 10);
✵ инактивации метаболитов (гормонов, медиаторов и др.) и обезвреживания чужеродных соединений, включая и лекарственные препараты (см. подразд. IX, раздел 12).
Регенерация метионина
Реакции метилирования играют важную роль в организме и протекают очень интенсивно. Это вызывает большой расход метионина, так как он является незаменимой аминокислотой (в клетках метионин синтезироваться не может). В связи с этим большое значение приобретает возможность регенерации метионина с участием заменимых аминокислот (Сер, Гли). В результате отщепления метильной группы SAM превращается в S-аденозилгомоцистеин (SAT), который при действии гидролазы расщепляется на аденозин и гомоцистеин.
S-аденозилгомоцистеин + Н2O —> Аденозин + Гомоцистеин
Гомоцистеин может снова превращаться в метионин под действием гомоцистеинметилтрансферазы. Донором метильной группы в этом случае служит N5-метил-Н4-фолат:

Промежуточным переносчиком метильной группы в этой реакции служит производное витамина В12 — метилкобаламин, выполняющий роль кофермента.
Метионин — незаменимая аминокислота, однако может регенерироваться из гомоцистеина. Следовательно, незаменим именно гомоцистеин, но единственным его источником в организме служит метионин. В пище гомоцистеина крайне мало, поэтому потребности человека в метионине и гомоцистеине обеспечиваются только метионином пищи. Общая схема метаболизма метионина, связанная с обменом одноуглеродных фрагментов, представлена на рис. 9-27.
Рис. 9-27. Метаболизм метионина. 1 — реакции трансметилирования; 2 — синтез цистеина; 3 — регенерация метионина.

Первичным донором одноуглеродных фрагментов является серин. Образовавшийся N5, N10- метилен-Н4-фолат восстанавливается до N5-метил-Н4-фолата, передающего метальную группу на кобаламин (витамин В12). Метилкобаламин непосредственно участвует в регенерации метионина. Гомоцистеин может использоваться также для синтеза цистеина.
2. Обмен цистеина
Вторая серосодержащая аминокислота — цистеин. Она условно заменима, так как для её синтеза необходим атом серы, источником которого служит незаменимая аминокислота метионин.
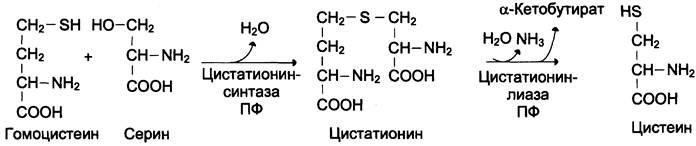
Для синтеза цистеина необходимы 2 аминокислоты:
Серин — источник углеродного скелета;
Метионин — первичный источник атома Б (см. схему А).

Синтез цистеина из гомоцистеина происходит в 2 стадии под действием пиридоксальзависимых ферментов цистатионинсинтазы и цис- татионинлиазы (см. схему Б).
При нарушении использования гомоцистеина в организме из него образуется гомоцистин:
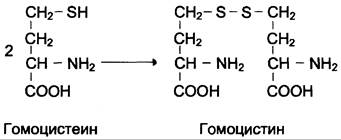
Гомоцистин может накапливаться в крови и тканях, выделяться с мочой, вызывая гомоцистинурию. Возможной причиной является наследственное нарушение обмена гомоцистеина либо гиповитаминоз фолиевой кислоты, а также витаминов В12 и В6. Из других биохимических нарушений можно отметить цистатионинурию, также часто возникающую при недостаточности витаминов группы В.
Биологические функции цистеина разнообразны и очень важны для организма. Так, цистеин, входящий в состав белков, играет необычайно важную роль в их фолдинге, поскольку тиогруппы цис способны образовывать прочную дисульфидную связь. При этом 2 остатка цистеина формируют молекулу цистина (см. схему В).
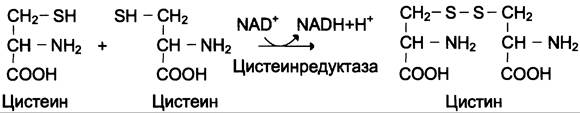
Окислительная реакция протекает либо с участием кофермента NАD+ под действием фермента цистеинредуктазы, либо неферментативно. Дисульфидные связи стабилизируют пространственную структуру полипептидной цепи или связывают между собой 2 цепи (например, А- и В-цепи гормона инсулина). Очень многие белки и ферменты в активном центре содержат SН- группы, участвующие в катализе. При их окислении ферментативная активность падает (см. разделы 1, 2). Восстановление SН-групп часто происходит с использованием глутатиона — атипичного трипептида, содержащего y-глутаминовую кислоту, цистеин и глицин (см. схему Г).

Глутатион способен существовать в 2 формах — восстановленной (Г-SН) и окисленной (Г-S-S-Г) и служит активным антиоксидантом в организме человека.
Ещё одним важным путём использования цистеина можно считать синтез таурина в животных тканях, который происходит путём декарбоксилирования производных цистеина — цистеиновой и цистеинсульфиновой кислот:
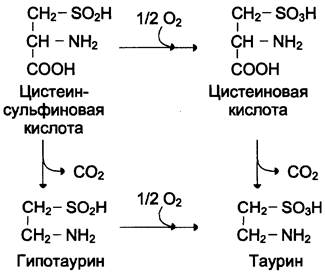
Таурин необходим для синтеза парных жёлчных кислот в печени. Кроме того, он очень важен в клетках как антиоксидант и используется для снижения ПОЛ и связывания гипохлорит- аниона (в форме хлораминового комплекса).
Цистеин также служит предшественником тиоэтаноламинового фрагмента HS-KoA (кофермента А).
Катаболизм цистеина происходит окислительным путём (см. схему А).
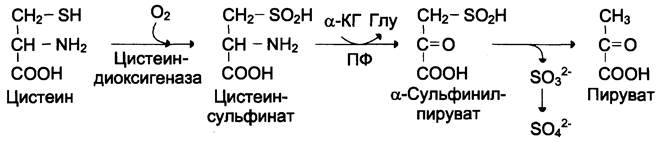
Сульфит, который получается в реакции, превращается в сульфат и выводится с мочой, либо превращается в эфиро-серные кислоты, которые также экскретируются почками. Цистеин — практически единственный источник сульфатов мочи.
Пути использования цистеина представлены на схеме (см. схему Б).

В. Метаболизм фенилаланина и тирозина
Фенилаланин — незаменимая аминокислота, так как в клетках животных не синтезируется её бензольное кольцо. Тирозин — условно заменимая аминокислота, поскольку образуется из фенилаланина. Содержание этих аминокислот в пищевых белках (в том числе и растительных) достаточно велико. Фенилаланин и тирозин используются для синтеза многих биологически активных соединений. В разных тканях метаболизм этих аминокислот происходит по-разному (рис. 9-28).
Рис. 9-28. Пути превращения фенилаланина и тирозина в разных тканях. Н4БП — тетрагидробиоптерин; Н2БП — дигидроби- оптерин; ПФ — пиридоксальфосфат; SАМ — S-аденозил метионин.

1. Метаболизм фенилаланина
Основное количество фенилаланина расходуется по 2 путям:
✵ включается в белки;
✵ превращается в тирозин.
Превращение фенилаланина в тирозин прежде всего необходимо для удаления избытка фенилаланина, так как высокие концентрации его токсичны для клеток. Образование тирозина не имеет большого значения, так как недостатка этой аминокислоты в клетках практически не бывает.
Основной путь метаболизма фенилаланина начинается с его гидроксилирования (рис. 9-29), в результате чего образуется тирозин. Эта реакция катализируется специфической монооксигеназой — фенилаланингидроксилазой, коферментом которой служит тетрагидробиоптерин (Н4БП). Активность фермента зависит также от наличия Fе2+. Реакция необратима. Н4БП в результате реакции окисляется в дигидробиоптерин (Н2БП). Регенерация последнего происходит при участии дигидроптеридинредуктазы с использованием NАDРН + Н+.
Рис. 9-29. Реакции гидроксилирования фенилаланина (1) и регенерации Н4БП (2).

2. Особенности обмена тирозина в разных тканях
Обмен тирозина значительно сложнее, чем обмен фенилаланина. Кроме использования в синтезе белков, тирозин в разных тканях выступает предшественником таких соединений, как катехоламины, тироксин, меланины, и катаболизируется до СO2 и Н2O.
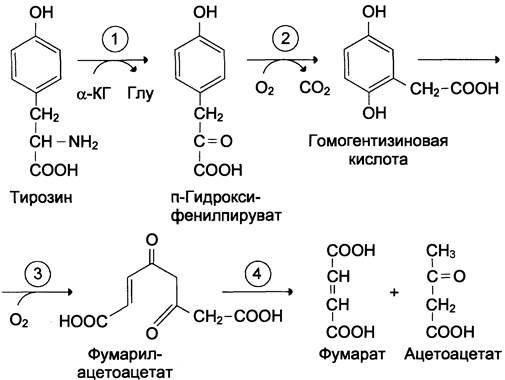
Катаболизм тирозина в печени
В печени происходит катаболизм тирозина до конечных продуктов. Специфический путь катаболизма включает несколько ферментативных реакций, завершающихся образованием фумарата и ацетоацетата (см. схему А):
1. Трансаминирование тирозина с α-кетоглутаратом катализирует тирозинаминотрансфераза (кофермент ПФ) — индуцируемый фермент печени млекопитающих. В результате образуется п-гидроксифенилпируват.
2. В реакции окисления п-гидроксифенилпирувата в гомогентизиновую кислоту происходит декарбоксилирование, гидроксилирование ароматического кольца и миграция боковой цепи. Реакцию катализирует фермент п-гидроксифенилпируватдиоксигеназа, кофакторами которого выступают витамин С и Fе2+.
3. Превращение гомогентизиновой кислоты в фумарилацетоацетат сопровождается расщеплением ароматического кольца. Эта реакция катализируется диоксигеназой гомогентизиновой кислоты, в качестве кофермента содержащей Fе2+.
Обмен фенилаланина и тирозина связан со значительным количеством реакций гидроксилирования, которые катализируют оксигеназы. Ферменты оксигеназы (гидроксилазы) используют молекулу O2 и кофермент-донор водорода (чаще — Н4БП). Для катализа оксигеназам необходимы кофакторы — Fе2+ или гем (для некоторых — Cu+), а для многих ещё и витамин С. Оксигеназы делят на 2 группы:
Монооксигеназы — один атом О2 присоединяют к продукту реакции, другой используют для образования Н2O;
Диоксигеназы — оба атома O2 используют для образования продукта реакции.
Почти все процессы расщепления ароматических колец в биологических системах катализируются диоксигеназами, подклассом ферментов, открытым японским биохимиком Осами Хайяши.
В результате разрыва бензольного кольца образуется малеилацетоацетат, который в процессе цис- и транс-изомеризации превращается в фумарил ацетоацетат.
4. Гидролиз фумарил ацетоацетата при действии фумарилацетоацетатгидролазы приводит к образованию фумарата и ацетоацетата. Фумарат может окисляться до СO2 и Н2O или использоваться для глюконеогенеза. Ацетоацетат — кетоновое тело, окисляемое до конечных продуктов с выделением энергии.
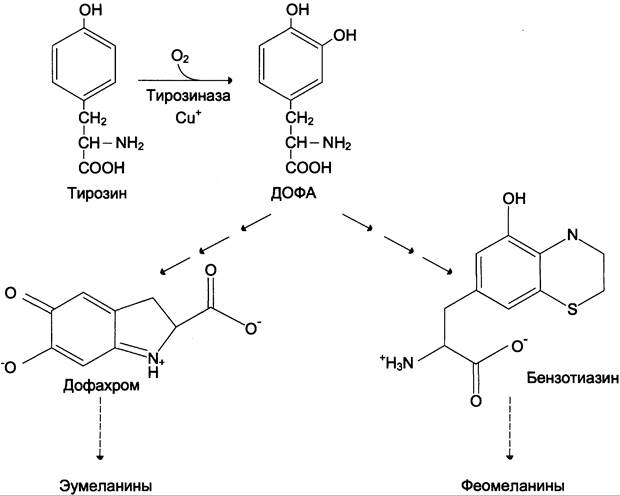
Превращение тирозина в меланоцитах
В пигментных клетках (меланоцитах) тирозин выступает предшественником тёмных пигментов — меланинов. Среди них преобладают 2 типа: эумеланины и феомеланины. Эумелани- ны (чёрного и коричневого цвета) — нерастворимые высокомолекулярные гетерополимеры 5,6-дигидроксииндола и некоторых его предшественников. Феомеланины — жёлтые или красновато-коричневые полимеры, растворимые в разбавленных щелочах. Находятся они, в основном, в составе волос. Меланины присутствуют в сетчатке глаз. Цвет кожи зависит от распределения меланоцитов и количества в них разных типов меланинов.
Синтез меланинов — сложный, многоступенчатый, разветвлённый процесс. Краткая схема синтеза представлена на рис. 9-28.
Первую реакцию — превращение тирозина в ДОФА — катализирует тирозиназа, использующая в качестве кофактора ионы Сu+ (см. схему А).
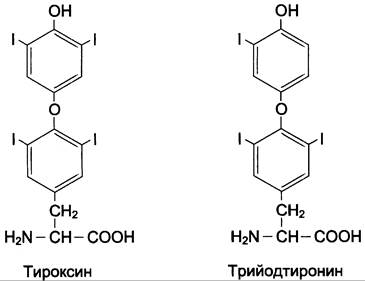
Превращение тирозина в щитовидной железе
В щитовидной железе синтезируются и выделяются гормоны йодтиронины: тироксин (тетрайодтиронин) и трийодтиронин. Эти гормоны представляют собой йодированные остатки тирозина, которые попадают в клетки щитовидной железы через базальную мембрану (см. раздел 11).
Превращения тирозина в надпочечниках и нервной ткани (синтез катехоламинов)
В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов (дофамина, норадреналина и адреналина) (см. схему Б).
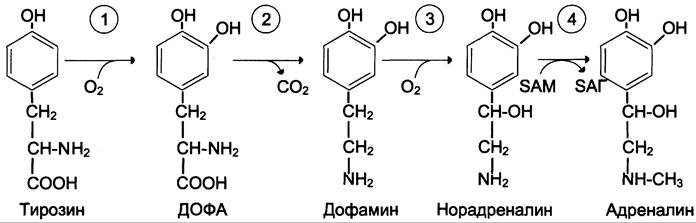
При образовании катехоламинов, которое происходит в нервной ткани и надпочечниках, и меланина в меланоцитах промежуточным продуктом служит диоксифенилаланин (ДОФА). Однако гидроксилирование тирозина в клетках различных типов катализируется различными ферментами:
Тирозиназа в меланоцитах является Сu+-зависимым ферментом (см. выше).
Тирозингидроксилаза (1) в надпочечниках и катехоламинергических нейронах не нуждается в ионах меди. Это — Fe2+-зависимый фермент, аналогично фенилаланингидроксилазе в качестве кофермента использующий Н4БП. Физиологическая роль тирозингидроксилазы чрезвычайно велика, так как этот фермент является регуляторным и определяет скорость синтеза катехоламинов.
Активность тирозингидроксилазы значительно изменяется в результате: Аллостерической регуляции (ингибитор — норадреналин);
Фосфорилирования/дефосфорилирования: в результате фосфорилирования с участием протеинкиназы А снижаются Кm для кофермента Н4БП и сродство фермента к норадреналину, в результате чего происходит активация тирозингидроксилазы.
Количество фермента регулируется на уровне транскрипции.
ДОФА-декарбоксилаза (2) (кофермент — ПФ) катализирует образование дофамина, который при участии дофамингидроксилазы (3) (монооксигеназы) превращается в норадреналин. Для функционирования фермента необходимы ионы Сu+, витамин С и тетрагидробиоптерин.
В мозговом веществе надпочечников фенил- этаноламин- N-метилтрансфераза (4) катализирует метилирование норадреналина, в результате чего образуется адреналин. Источником метильной группы служит SAM.
Дофамин и норадреналин служат медиаторами в синаптической передаче нервных импульсов, а адреналин — гормон широкого спектра действия, регулирующий энергетический обмен. Одна из функций катехоламинов — регуляция деятельности ССС (см. раздел 11).
3. Заболевания, связанные с нарушением обмена фенилаланина и тирозина
Известно несколько наследственных заболеваний, связанных с дефектом ферментов обмена фенилаланина и тирозина в разных тканях.
Фенилкетонурия
В печени здоровых людей небольшая часть фенилаланина (~10%) превращается в фенил- лактат и фенилацетилглутамин (рис. 9-30).
Рис. 9-30. Альтернативные пути катаболизма фенилаланина. При дефекте фенилаланингидроксилазы накопившийся фенилаланин подвергается трансаминированию с α-кетоглутаратом. Образовавшийся фенилпируват превращается либо в фениллактат, либо в фенилацетилглутамин, которые накапливаются в крови и выделяются с мочой. Эти соединения токсичны для клеток мозга.

Этот путь катаболизма фенилаланина становится главным при нарушении основного пути — превращения в тирозин, катализируемого фенилаланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглутамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ:
Классическая ФКУ — наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20 — 30 раз (в норме — 1,0 — 2,0 мг/дл), в моче — в 100 — 300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300 — 600 мг/дл при полном отсутствии в норме.
Наиболее тяжёлые проявления ФКУ — нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания — 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу.
Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие концентрации фенилаланина ограничивают транспорт тирозина и триптофана через гематоэнцефалический барьер и тормозят синтез нейро- медиаторов (дофамина, норадреналина, серотонина).
Вариантная ФКУ (коферментзависимая ги- перфенилаланинемия) — следствие мутаций в генах, контролирующих метаболизм Н4БП. Клинические проявления — близкие, но не точно совпадающие с проявлениями классической ФКУ. Частота заболевания — 1 — 2 случая на 1 млн новорождённых.
Н4БП необходим для реакций гидроксилирования не только фенилаланина, но также тирозина и триптофана, поэтому при недостатке этого кофермента нарушается метаболизм всех 3 аминокислот, в том числе и синтез нейромедиаторов. Заболевание характеризуется тяжёлыми неврологическими нарушениями и ранней смертью («злокачественная» ФКУ).
Прогрессирующее нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина. Если такое лечение начато сразу после рождения ребёнка, то повреждение мозга предотвращается. Считается, что ограничения в питании могут быть ослаблены после 10-летнего возраста (окончание процессов миелинизации мозга), однако в настоящее время многие педиатры склоняются в сторону «пожизненной диеты».
Для диагностики ФКУ используют качественные и количественные методы обнаружения патологических метаболитов в моче, определение концентрации фенилаланина в крови и моче. Дефектный ген, ответственный за фенилкетонурию, можно обнаружить у фенотипически нормальных гетерозиготных носителей с помощью теста толерантности к фенилаланину. Для этого обследуемому дают натощак ~10 г фенилаланина в виде раствора, затем через часовые интервалы берут пробы крови, в которых определяют содержание тирозина. В норме концентрация тирозина в крови после фенилаланиновой нагрузки значительно выше, чем у гетерозиготных носителей гена фенилкетонурии. Этот тест используется в генетической консультации для определения риска рождения больного ребёнка. Разработана схема скрининга для выявления новорождённых детей с ФКУ. Чувствительность теста практически достигает 100%.
В настоящее время диагностику мутантного гена, ответственного за ФКУ, можно проводить с помощью методов ДНК-диагностики (рестрикционного анализа и ПЦР).
Тирозинемии
Некоторые нарушения катаболизма тирозина в печени приводят к тирозинемии и тирози- нурии. Различают 3 типа тирозинемии.
Тирозинемия типа 1 (тирозиноз). Причиной заболевания является, вероятно, дефект фермента фумарилацетоацетатгидролазы, катализирующего расщепление фумарилацетоацетата на фумарат и ацетоацетат (рис. 9-28). Накапливающиеся метаболиты снижают активность некоторых ферментов и транспортных систем аминокислот. Патофизиология этого нарушения достаточно сложна. Острая форма тирозиноза характерна для новорождённых. Клинические проявления — диарея, рвота, задержка в развитии. Без лечения дети погибают в возрасте 6 — 8 мес из-за развивающейся недостаточности печени. Хроническая форма характеризуется сходными, но менее выраженными симптомами. Гибель наступает в возрасте 10 лет. Содержание тирозина в крови у больных в несколько раз превышает норму. Для лечения используют диету с пониженным содержанием тирозина и фенилаланина.
Тирозинемия типа II (синдром Рихнера—Ханхорта). Причина — дефект фермента тирозина- минотрансферазы. Концентрация тирозина в крови больных повышена. Для заболевания характерны поражения глаз и кожи, умеренная умственная отсталость, нарушение координации движений.
Тирозинемия новорождённых (кратковременная). Заболевание возникает в результате снижения активности фермента п - гидроксифенил- пируватдиоксигеназы, превращающего п-гидроксифенилпируват в гомогентизиновую кислоту (рис. 9-28). В результате в крови больных повышается концентрация п-гидроксифенилацетата, тирозина и фенил-аланина. При лечении назначают бедную белком диету и витамин С.
Алкаптонурия («чёрная моча»)
Причина заболевания — дефект диоксигеназы гомогентизиновой кислоты (рис. 9-28). Для этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Это метаболическое нарушение было описано ещё в XVI веке, а само заболевание охарактеризовано в 1859 г. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота — 2 — 5 случаев на 1 млн новорождённых. Заболевание наследуется по аутосомно-рецессивному типу. Диагностических методов выявления гетерозиготных носителей дефектного гена к настоящему времени не найдено.
Альбинизм
Причина метаболического нарушения — врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в меланоцитах. В результате дефекта тирозиназы нарушается синтез пигментов меланинов.
Клиническое проявление альбинизма (от лат. albus — белый) — отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. Частота заболевания 1:20 000.
Нарушение синтеза катехоламинов (рис. 9-28) может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества.
Болезнь Паркинсона
Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. Это одно из самых распространённых неврологических заболеваний (частота 1:200 среди людей старше 60 лет). При этой патологии снижена активность тирозингидроксилазы, ДОФА-декарбоксилазы. Заболевание сопровождается тремя основными симптомами: акинезия (скованность движений), ригидность (напряжение мышц), тремор (непроизвольное дрожание). Дофамин не проникает через гематоэнцефалический барьер и как лекарственный препарат не используется. Для лечения паркинсонизма предлагаются следующие принципы:
• заместительная терапия препаратами-предшественниками дофамина (производными ДОФА) — леводопа, мадопар, наком и др.
• подавление инактивации дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.).
Депрессивные состояния часто связаны со снижением в нервных клетках содержания дофамина и норадреналина.
Гиперсекреция дофамина в височной доле мозга наблюдается при шизофрении.