БИОХИМИЯ И МОЛЕКУЛЯРНАЯ БИОЛОГИЯ - В. ЭЛЛИОТ - 2002
ГЛАВА 1. ХИМИЯ, ЭНЕРГИЯ И МЕТАБОЛИЗМ
Жизнь - это химический процесс, включающий тысячи упорядоченно протекающих реакций. Их называют метаболическими реакциями или, обобщенно, метаболизмом.
Поэтому, изучая биохимию, вы встретитесь с множеством (но все же не с тысячами!) химических уравнений, описывающих наиболее существенные аспекты химии клетки. Биохимические стратегии, отобранные и отшлифованные за миллионы лет эволюции, элегантны и покоряюще красивы, но, чтобы оценить их по достоинству, понимать и восхищаться ими, сначала нужно понять, с какими проблемами связано само существование жизни.
Анализ любого биологического превращения основан на представлении об энергии. Записать его в виде химического уравнения недостаточно, если при этом нет сведений о происходящих изменениях химической энергии. Без этого нельзя понять, сможет ли это превращение сколько-нибудь заметно протекать в растворе и в какой мере оно будет уравновешиваться обратной реакцией.
Насколько важнее такой подход, можно видеть на следующем примере. При интенсивной работе мышц полисахарид гликоген (полимер глюкозы) в серии химических реакций превращается в молочную кислоту, а при последующем отдыхе он вновь синтезируется из молочной кислоты, причем не в результате простого обращения реакции, а иным путем. Простые энергетические соображения позволяют понять, почему клетка поступает именно так.
Для понимания биохимических процессов достаточно самых общих знаний термодинамики. Есть, разумеется, области, где этого недостаточно, но в целом большинству биохимиков хватает знаний основных принципов, и они редко используют термодинамические уравнения. Этот раздел призван обеспечить именно такую степень понимания.
Что определяет возможность протекания химических реакций?
Как уже отмечалось, только изменение энергии определяет, какие химические реакции возможны, а какие нет. Заметим, что в биохимии нас интересуют реакции, протекающие с образованием большого количества продукта. В принципе, термодинамические факторы не препятствуют полному протеканию реакций (за исключением случаев, когда субстраты и продукты находятся в равновесных концентрациях; см. ниже). Но если реакция останавливается после того, как произошло минимальное превращение субстрата, то с точки зрения жизни такая реакция не имеет значения.
Сначала определим, что имеется в виду под изменением энергии химической системы, т. е. ансамбля молекул, участвующих в химических реакциях. Это не так легко понять, как, например, изменение гравитационной энергии при падении тела. Химическая система состоит из огромного числа отдельных молекул, в каждой из которых заключено некоторое количество энергии, определяемое ее структурой. Эта энергия может быть представлена как теплосодержание, или энтальпия, молекулы. Когда структура молекулы меняется в ходе химической реакции, изменение её энергии описывается как изменение энтальпии АН. Оно может быть отрицательным (теплота теряется молекулами и рассеивается, увеличивая температуру окружающей среды) или положительным (теплота поглощается из окружающей среды, которая при этом охлаждается).
На первый взгляд может показаться, что реакции с положительным АН столь же нереальны, как тело, самопроизвольно взлетающее к потолку. Но такая простая физическая аналогия неприменима к химическим реакциям: здесь знак изменения энтальпии не определяет жестко направление процесса, а лишь показывает, что этот фактор либо способствует ему, либо препятствует. Куда будет падать тело, зависит только от изменения гравитационной энергии, а какая пойдет реакция, зависит не только от изменения энтальпии, но также и от изменения энтропии (∆S) химической системы.
Энтропия может быть определена как степень неупорядоченности. В химической системе беспорядок проявляется по-разному. Поскольку молекула обычно не жесткая структура, она может вибрировать, вращаться как единое целое и относительно отдельных своих связей. Чем легче происходит любое молекулярное движение, тем больше беспорядка, или энтропии. Кроме того, огромное число молекул, образующих химическую систему, может быть случайным образом рассеяно в пространстве или существовать в нем в той или иной мере упорядочено, как это имеет место в живых клетках. И наконец, число отдельных молекул и ионов в системе может измениться в результате химических реакций. Чем больше молекул, тем больше их совокупная неупорядоченность, а, следовательно, и энтропия системы.
И ∆Н, и ∆S имеют право голоса при решении вопроса, пойдет ли химическая реакция. Отрицательное ∆Н и положительное ∆S вместе дают ответ «да», положительное ∆Н и отрицательное ∆S — ответ «нет». Если же изменения энтальпии и энтропии имеют одинаковые знаки, их влияние противоположно, и вопрос решается сравнением их величин.
Рост энтропии как движущей силы процесса можно проиллюстрировать на примере таяния льда в теплой воде. Плавление льда происходит с поглощением тепла (∆Н- положительно), однако увеличение молекулярного беспорядка при разрушении кристаллической решетки столь велико, что именно оно является определяющим фактором процесса. Гораздо сложнее представить, как энтропия управляет химической реакцией. Общее утверждение состоит в том, что при сопоставимых значениях энергии предпочтительнее более неупорядоченное состояние системы. Если вам трудно это понять - примите пока на веру, далее мы к этому еще вернемся.
Сравнивать изменение энтальпии и энтропии, которые могут быть союзниками или антагонистами, при оценке возможности протекания реакции крайне неудобно; хотя бы потому, что их величины имеют разную размерность. Более того, в биологических системах прямое измерение энтропии затруднительно или невозможно. Ситуация, однако, значительно улучшается благодаря предложенному Гиббсом понятию свободной энергии, которое объединяет оба рассмотренных выше понятия - энтальпию и энтропию. Изменение свободной энергии (следуя Гиббсу, обозначим его ∆G) описывает знаменитое уравнение:
∆G = ∆Н - Т∆S,
в котором Т- абсолютная температура.
Определение свободная здесь означает не свободу вообще, а свободу использовать эту энергию для совершения полезной работы. ∆G представляет собой максимальное значение энергии, которое доступно для совершения полезной работы за счет химической реакции. Иначе говоря, это та часть ваших денег, на которую вы можете рассчитывать при покупках.
Применительно к биологии, полезная работа-это мышечное сокращение, химический синтез в клетках, преодоление осмотических или электрических сил. Величину ∆Gвыражают в калориях (кал) или, что более принято, в джоулях (Дж) на моль (1 кал = 4,19 Дж). Поскольку значения ∆G обычно велики, чаще пользуются килокалориями или килоджоулями.
Изменение свободной энергии в ходе любого процесса - важнейший термодинамический параметр. Применительно к химическим процессам можно сформулировать общее правило: химическая реакция протекает лишь в случае ∆G <0, т. е. есть в условиях, когда свободная энергия продуктов реакции меньше, чем исходных веществ.
Изменение свободной энергии и обратимость реакций
Вероятно, вам уже говорили, что многие химические реакции обратимы. Это может озадачить, ведь тогда получается, что величины ∆G отрицательны и для прямой, и для обратной реакции (вспомним, что это критерий возможности спонтанного протекания любого процесса). Разгадка кажущегося парадокса состоит в том, что ∆G не является фиксированным параметром и зависит от концентраций исходных веществ и продуктов их превращения.
С уравнением, описывающим эту зависимость, мы познакомимся позже. А пока заметим, что для обратимой реакции А <-> В изменение свободной энергии ∆G может быть отрицательным применительно к превращению А —> В и положительным применительно к превращению В —> А, если только концентрация вещества А больше концентрации вещества В. При обратном соотношении концентраций А и В отрицательным может стать изменение свободной энергии ∆G при превращении В —> А.Нетрудно сообразить, что существует некоторое соотношение концентраций, при котором изменения ∆G для прямой и обратной реакции равны нулю. Такое соотношение будет сохраняться сколь угодно долго - это точка химического равновесия.
Если величина ∆G для некоторой биохимической реакции А <-> В невелика по модулю, то с большей вероятностью эта реакция в клетке будет обратима, поскольку изменение концентрации веществ А и В в ходе метаболизма может привести к обращению знака ∆G. Напротив, если абсолютное значение ∆G велико, ре- акцию можно считать практически необратимой. Диапазон изменений внутриклеточных концентраций метаболитов (термин, объединяющий всех участников биохимических реакций) относительно узок: обычно значения концентраций лежат в пределах 10-4-10-3 М. В результате реакции с большими по модулю отрицательными значениями ∆G реально необратимы, так как концентрационных изменений недостаточно для обращения знака ∆G.
Как правило, внутриклеточный гидролиз, т. е. реакции расщепления связей с участием молекулы воды, необратим в том смысле, что образование этих связей в клетках не является результатом обращения гидролиза. О том, насколько обратимы другие внутриклеточные реакции, будет рассказано в главе о метаболизме.
Итак, внутриклеточные реакции, сопровождающиеся незначительным изменением свободной энергии, могут быть обратимы, причем их направление зависит от небольших изменений концентраций участвующих метаболитов. Напротив, при значительном изменении свободной энергии метаболические реакции идут в одном направлении и реально протекают до полного завершения, поскольку именно в эту область концентраций сдвинута точка равновесия. Иными словами, исходные вещества полностью превращаются в продукты реакции.
Роль необратимых реакций в стратегии метаболизма
В предыдущем разделе мы подчеркнули влияние величины ∆G химической реакции на возможность ее протекания в обратном направлении. Пора объяснить, почему это так важно. Основные физиологически важные внутриклеточные химические процессы обычно включают не одну, а много последовательно протекающих реакций, объединенных в так называемые метаболические пути, где продукт первой реакции является исходным веществом для второй и т. д. Например, в упомянутом выше превращении гликогена в молочную кислоту в мышцах вовлечено двенадцать последовательных химических реакций.
Общим свойством метаболических путей является их необратимость. Это не означает, что необратимы все включенные в них реакции. Но по меньшей мере одна реакция в физиологических условиях не протекает в обратном направлении. Такие реакции, действуя подобно клапанам на трубопроводе, и обеспечивают термодинамическую основу завершенности суммарного метаболического превращения.
Это, однако, не означает физиологической необратимости результирующего превращения. Так, из молочной кислоты в организме благополучно синтезируется гликоген.
Некоторые из стадий биосинтеза гликогена представляют собой обращенные реакции его расщепления. Но есть другие реакции, которые не могут быть обращены, и вместо них используются альтернативные пути. Например, некоторые реакции протекают с использованием энергии, что делает их необратимыми в обратном направлении. В результате и прямой (гликоген —> молочная кислота), и обратный (молочная кислота —> гликоген) метаболические пути являются необратимыми. Типичная метаболическая ситуация может быть описана следующей схемой:
![]()
На этой схеме красными стрелками помечены необратимые реакции с большими по модулю отрицательными значениями ∆G. Чуть позже мы расскажем, как достигается энергетическое обеспечение необратимости в обоих направлениях.
Итак, стал ясен общий биохимический принцип. Всякий раз, когда суммарный химический процесс, характерный для данного метаболического пути, должен быть физиологически обратимым, прямая и обратная траектории не совпадают полностью и обязательно содержат хотя бы пару различающихся по своей природе необратимых реакций.
Почему клетки используют такую стратегию метаболизма?
Тому есть две основные причины. Рассмотрим альтернативную стратегию, при которой все метаболические реакции обратимы.
![]()
Главный недостаток при этом - подверженность процесса в целом закону действующих масс. Если концентрация вещества А возрастает (скажем, вы что-нибудь съели), равновесие сдвигается вправо и возрастает концентрация конечных продуктов. Напротив, при уменьшении концентрации А некоторые продукты будут превращаться в исходные вещества. Представьте теперь, что речь идет о биосинтезе ДНК генов или жизненно важных белков. И вы поймете, насколько неприемлем такой сценарий.
Есть и вторая сходная причина, по которой прямое и обратное направления метаболического пути должны включать различные реакции: метаболизм обязан быть контролируемым. Мы уже знаем, что при интенсивной работе мышц гликоген превращается в молочную кислоту, тогда как при отдыхе этот процесс выключается и происходит ресинтез гликогена из молочной кислоты. Чтобы независимо управлять обоими процессами, т. е. включать один и выключать другой, они должны различаться. Иначе их можно включать и выключать только одновременно! Как правило, именно необратимые стадии метаболизма являются местом приложения регуляторных механизмов. Подробнее об этом в главе 12.
Из чего складывается величина ∆G?
Хотя изменение свободной энергии при химической реакции, как уже отмечалось, непостоянно, в некоторых определенных условиях, принятых за стандартные, величина ∆Gявляется постоянной. Примем в качестве таких стандартных условий концентрации всех веществ-участников 1,0 М, температуру 25° С и значение pH 7,0. Отвечающую этим условиям величину ∆G называют стандартным изменением свободной энергии данной реакции и обозначают ∆G°′. Заметим, что некоторые физики и химики предпочитают в качестве стандартного условия значение pH 1,0, вместо 7,0.
Для вычисления стандартного изменения свободной энергии различных реакций часто используют физико-химические таблицы, в которых приведены стандартные свободные энергии образования большого числа химических соединений. Если по отдельности сложить значения для исходных веществ и продуктов реакции, то разность между этими двумя суммами и будет искомой величиной ∆G°′. Альтернативой является экспериментальное определение константы равновесия данной реакции, из которой нетрудно вычислить ∆G°′.
При заданных концентрациях исходных веществ и продуктов реакции величины ∆G и ∆G°′ связывает простое соотношение. Поэтому, зная внутриклеточные концентрации метаболитов, можно вычислить изменение свободной энергии, характеризующее соответствующую биохимическую реакцию. Это было проделано для очень многих биохимических реакций, и на полученные таким образом значения часто ссылаются.
Однако совсем не просто определить внутри клеток концентрации тысяч метаболитов, содержание которых там мало и непостоянно. И потому для многих биохимических реакций значения ∆G пока остаются неизвестными. К счастью, оказалось, что во многих случаях можно использовать величины ∆G°′, хотя отвечающие им условия в клетках никогда не реализуются. Разумеется, это компромисс, но полезный.
Стандартная свободная энергия и константы равновесия
Знать величины ∆G°′ особенно полезно, потому что из них легко вычисляется константа равновесия соответствующей реакции. Константа равновесия К′eq представляет собой отношение произведений равновесных концентраций продуктов реакции и исходных веществ (штрих означает, что константа равновесия отвечает pH 7,0). Так, например, для реакции А + В <-> С + D константа Кeq вычисляется из концентраций веществ А, В, С и D, измеренных в условиях равновесия, т. е. когда эти концентрации перестали изменяться.
![]()
Связь между величинами ∆G°′ и К′еq описывается уравнением:
∆G°′ = RТ • In К′eq = -2,303RТ • Ig К'еq,
где R - газовая постоянная (8,315 кДж • моль-1 • К-1); Т - абсолютная температура в градусах Кельвина (298 К - 25° С). При 25° С RТ = 2,478 кДж • моль-1. Таким образом, измерив значение К’eq, можно вычислить величину ∆G°′, и наоборот, располагая сведениями о ∆G°′, можно вычислить значение константы равновесия К'eq.
Таблица 1.1. Соотношение между константой равновесия реакции (K'eq) и изменением свободной энергии (∆G°′)
Таблица позволяет составить представление о соотношении констант равновесия химических реакций со стандартными изменениями свободной энергии
∆G°′ (кДж • моль-1) |
К'еq |
+ 17,1 |
0,001 |
+ 11,4 |
0,01 |
+5,7 |
0,1 |
0 |
1,0 |
-5,7 |
10,0 |
-11,4 |
100,0 |
-17,1 |
1000,0 |
Допустим, что ∆G отрицательно. От чего зависит, будет ли протекать соответствующая реакция в клетке с ощутимой скоростью?
Для протекания химической реакции необходимо, чтобы ей отвечало уменьшение свободной энергии. Однако из этого вовсе не следует, что она будет протекать с ощутимой скоростью. Оценка с точки зрения термодинамики реакции взаимодействия сахара с кислородом (т. е. горения) с образованием Н2O и СO2 показывает, что энергетически это очень выгодный процесс. В то же время сахар на воздухе вполне устойчив. Существует барьер для протекания реакций даже с очень большим уменьшением свободной энергии, иначе все горючие материалы на Земле давно бы исчезли в огне. Однако сахар, столь устойчивый в чашке с чаем, быстро окисляется в организме, как только мы выпиваем этот чай. Возникает вопрос: почему реакции, протекающие в живых клетках, вне их либо не идут вообще, либо идут с ничтожно малыми скоростями? Потому что в клетках они осуществляются с помощью ферментативного катализа.
Природа ферментативного катализа
В простейшем случае фермент (или энзим) - это катализатор, ускоряющий только одну химическую реакцию. Известны тысячи биохимических реакций, и каждая из них катализируется своим ферментом. Принцип «одна реакция - один фермент» соблюдается и в случае мультифункциональных ферментов, обладающих различными каталитическими активностями, и в случае мультиферментных комплексов.
Фермент является белком. И хотя строению белков будет посвящена глава 2, краткий экскурс в эту область здесь будет полезен. Белки построены из 20 аминокислот, соединенных в длинные цепи. Это высокомолекулярные соединения: даже у самых маленьких ферментов молекулярная масса порядка 10 000 дальтон (Да), а у многих она достигает сотен тысяч дальтон. Одна из причин, по которой ферменты так велики, заключается в том, что длинная цепь (или цепи), из которой они состоят, должна свернуться с образованием некого кармана, называемого активным центром. Попадая в такой карман, молекула вещества с исключительной точностью атакуется функциональными группами фермента. Под «атакой» здесь следует понимать химическое превращение вещества, которое принято называть субстратом, при участии данного фермента.
В целом ферментативный процесс обычно описывают моделью Михаэлиса-Ментен:
Е + S <-> ЕS <-> ЕР —> Е + Р,
где Е - фермент; S - субстрат; ЕS и ЕР - соответственно комплексы фермента с субстратом и продуктом ферментативной реакции (Р).
Из уравнения, описывающего эту модель, следует, что субстрат обратимо связывается с активным центром фермента и далее в связанном виде превращается в новое вещество, которое диффундирует из активного центра в окружающий раствор. Мы еще вернемся к модели Михаэлиса-Ментен при обсуждении регуляции ферментативного катализа (см.с. 156), а пока рассмотрим основы механизма катализа.
Как работает фермент?
Чтобы ответить на этот вопрос, нам следует понять природу химических реакций. Они протекают в две стадии. Рассмотрим превращение S —> Р.
На первой стадии исходная молекула претерпевает определенные конформационные и электронные изменения, которые служат предпосылкой ее последующего легкого преобразования в конечный продукт.
![]()
В таком возбужденном состоянии![]() (его называют переходным) молекула существует очень недолго (10-14- 10-13 с). Ясно, что возникновение переходного состояния обусловлено поступлением энергии извне.
(его называют переходным) молекула существует очень недолго (10-14- 10-13 с). Ясно, что возникновение переходного состояния обусловлено поступлением энергии извне.
Суммарная реакция S —> Р должна сопровождаться уменьшением свободной энергии, иначе она вообще бы не произошла. Этот термодинамический параметр зависит только от строения исходного и конечного соединений и инвариантен к характеру промежуточных изменений свободной энергии, который обычно называют энергетическим путем,или энергетическим профилем реакции. Поэтому нет никакого противоречия в том, что в переходном состоянии![]() молекула обладает большей свободной энергией, чем в исходном S. Положительное изменение свободной энергии при превращении S —>
молекула обладает большей свободной энергией, чем в исходном S. Положительное изменение свободной энергии при превращении S —>
называют энергией активации для данной реакции (рис. 1.1).
Рис. 1.1. Энергетические профили обычной и ферментативной реакций. Скорость реакции очень чувствительна к изменениям энергии активации, так как связана с ней обратной экспоненциальной зависимостью. S — субстрат; ![]() - переходное состояние; Р - продукт
- переходное состояние; Р - продукт
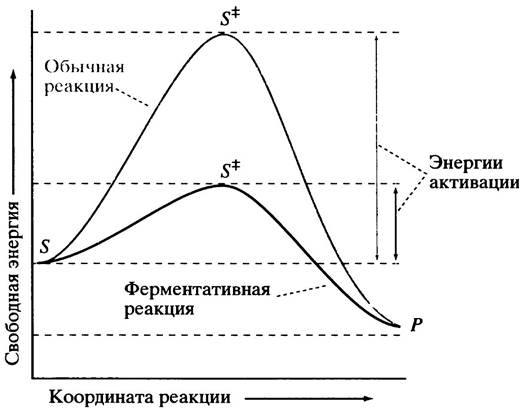
Энергетический горб (см. рис. 1.1) служит барьером на пути химической реакции. Без него энергетический профиль отражал бы непрерывное падение свободной энергии на пути от S к Р, и все вещества, способные вступать в реакции, сразу бы в них вступали.
Итак, чтобы реакция прошла, необходимо внести в систему энергию активации. В некатализируемых реакциях источником этой энергии служат столкновения между молекулами в растворе. Если соударяющиеся молекулы должным образом ориентированы, есть шанс, что возникающие при этом искажения молекулы Б будут такого свойства, что ее высокоэнергетическое состояние окажется переходным на пути S —> Р. Следовательно, скорость протекания реакции определяется вероятностью возникновения переходного состояния исходных молекул. С ростом температуры возрастает как скорость теплового движения, так и частота соударений, и потому увеличивается вероятность возникновения переходного состояния. Химики-органики издавна используют нагревание для ускорения реакций. Что же касается биохимических реакций, то в физиологическом диапазоне температур и в отсутствие катализаторов все они протекают с ничтожно малыми скоростями.
Дабы по достоинству оценить феномен ферментативного катализа, надо помнить, что для развития жизни был необходим такой способ быстрых химических превращений стабильных молекул, присутствующих в низких концентрациях в водных растворах с нейтральным pH, при котором реагирующие вещества достигали бы переходного состояния при физиологически низких температурах.
В активном центре каждого фермента есть по меньшей мере один участок, с которым могут связываться субстраты. В результате такого связывания молекула субстрата занимает наиболее выгодное положение для последующей химической реакции. Активный центр фермента полностью комплементарен молекуле субстрата в переходном состоянии. Непосредственно оценить прочность связывания фермента с субстратом, находящимся в переходном состоянии, нельзя. Но можно синтезировать такие аналоги субстрата, которые по своей конформации и распределению электронной плотности походили бы на переходное состояние. С подобными соединениями ферменты взаимодействуют с поразительно высоким сродством, иногда в тысячи раз большим, чем с собственным субстратом.
На это явление можно взглянуть и с других позиций, а именно с точки зрения перераспределения электронов. Аминокислотные остатки в активном центре устроены так и расположены таким образом, чтобы стабилизировать распределение электронов в переходном состоянии. Это означает, что активный центр лучше согласован с переходным состоянием, чем с субстратом. Иными словами, при связывании субстрата в нем возникают напряжения, снятие которых способствует возникновению переходного состояния. Суммарный эффект заключается в уменьшении энергии активации, т. е. приводит к снижению энергетического барьера на пути реакции (см. рис. 1.1). Однажды образовавшись, переходное состояние быстро превращается в продукты реакции со скоростью, не зависящей от структуры комплекса ![]() Эти продукты связаны с ферментом менее прочно и, высвобождаясь, диффундируют в окружающую среду. Скорость катализируемого процесса очень чувствительна к энергии активации. Даже столь малое ее изменение, которое отвечает образованию всего одной водородной связи (см. ниже), может ускорить реакцию в 106 раз. Ферменты ускоряют химические реакции обычно в 107-1014 раз. Например, уреаза, разрушающая мочевину, уменьшает энергию активации ее гидролиза на 84 кДж • моль-1 и ускоряет его в 1014 раз.
Эти продукты связаны с ферментом менее прочно и, высвобождаясь, диффундируют в окружающую среду. Скорость катализируемого процесса очень чувствительна к энергии активации. Даже столь малое ее изменение, которое отвечает образованию всего одной водородной связи (см. ниже), может ускорить реакцию в 106 раз. Ферменты ускоряют химические реакции обычно в 107-1014 раз. Например, уреаза, разрушающая мочевину, уменьшает энергию активации ее гидролиза на 84 кДж • моль-1 и ускоряет его в 1014 раз.
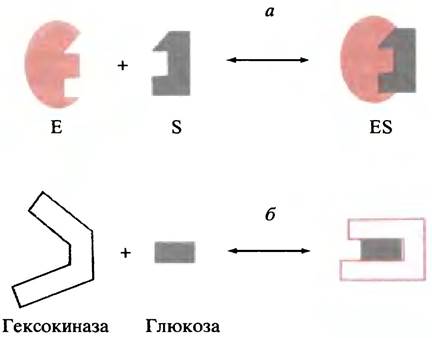
До сих пор мы описывали взаимодействие фермента с субстратом моделью «ключ-замок», в рамках которой субстрат должен быть комплементарен жесткому активному центру (рис. 1.2, а). Согласно современному представлению об индуцированном соответствии, активный центр фермента достаточно гибок и может изменять свою конформацию при связывании субстрата. Такого рода конформационные изменения были обнаружены, например, при рентгеноструктурных исследованиях гексокиназы, которая катализирует перенос фосфатного остатка с АТР на глюкозу. В структуре этого фермента можно выделить два «крыла», которые при связывании глюкозы «складываются», образуя каталитический центр (рис. 1.2, б). Этот конформационный переход определяет специфичность гексокиназы к глюкозе. Структура, которая связывает глюкозу, по-видимому, должна в какой-то степени связывать и другие полиолы и даже молекулы воды. В этом случае, исходя из модели «ключ-замок», можно ожидать от гексокиназы определенную активность в отношении других молекул, отличных от глюкозы. Применительно к воде это означало бы катализ гидролиза АТР, каковой в действительности не имеет места. Следовательно, каталитический центр возникает лишь при связывании «правильного» субстрата - глюкозы. Другие молекулы, если и связываются с ним, не способны вызвать те конформационные изменения фермента, которые обусловливают протекание ферментативной реакции.
Рис. 1.2. Взаимодействие субстрата (S) и фермента (Е) а - Модель «ключ-замок»; б - модель индуцированного соответствия
Большинство молекул могут вступать в различные химические реакции, каждой из которых отвечает свое переходное состояние. В отсутствие катализа, когда скорость превращения определяется лишь температурой, молекулы соударяются непредсказуемым образом, благодаря чему возникают самые разные переходные состояния. Поэтому основная реакция обычно сопровождается множеством побочных. Напротив, при ферментативном катализе ускоряется только одна из возможных реакций и образуются только определенные продукты превращения.
Особую роль ферменты могут играть в ускорении реакций посредством общего кислотно-основного катализа, когда принадлежащие активному центру протон-донорная или протон-акцепторная группы расположены таким образом, что непосредственно участвуют в переносе протона к переход ному состоянию связанного субстрата или от него. Точно так же (а иногда и одновременно) ферменты ускоряют реакции благодаря тому, что в их комплексах с субстратом «правильно» относительно переходного состояния расположен каталитически активный ион металла.
В ряде случаев ферменты выступают как участники химической реакции, образуя промежуточное ковалентное соединение с субстратом, которое, однако, далее распадается, высвобождая исходную молекулу белка. Примером здесь может служить важный класс ферментов - сериновые протеиназы, названные так потому, что их активные центры включают остаток аминокислоты серина, ковалентно участвующий в протеолизе:
R-CO-NHR-R' + Н2О —> RCOО- + R'NH3+.
В ходе этой каталитической реакции ацильный остаток, входящий в пептидную группу, сначала переносится на реакционноспособный гидроксил серина - ОН, а затем на молекулу воды
R-CO-NH-R- + Еnz-ОН + Н+ —> Еnz-О-СО-Р + NН3+-К, (1)
Еnz-О-СО-R + Н2O —> Еnz-ОН + R-COO- + Н+. (2)
Разные сериновые протеиназы специфичны к разным пептидам, но у всех каталитический центр содержит реакционноспособный остаток серина. Механизм действия этих ферментов детально изучен.
Скорость ферментативного катализа обычно является «визитной карточкой» данного фермента. В целом, все ферментативные реакции в клетке протекают неизмеримо быстрее некатализируемых, вклад которых попросту неощутим. Это ускорение, как уже отмечалось выше, может составлять 1014 раз и даже больше. Реально в ферментативные превращения вступает от нескольких до 107 молекул субстрата в секунду в расчете на одну молекулу фермента. Как будет показано далее (см. с. 157), скорость ферментативных реакций в определенном диапазоне зависит от концентрации субстрата. Если принять ее постоянной, то эта скорость будет определяться лишь внутриклеточным содержанием соответствующего фермента. Добавим, что многие ферменты обладают регуляторными механизмами, позволяющими изменять каталитическую активность в соответствии с физиологическими потребностями (см. главу 12, посвященную метаболическому контролю).
Активность фермента зависит от многих факторов. В их числе - степень ионизации его функциональных групп, которая определяет устойчивость белковой молекулы в целом и деятельность активного центра.
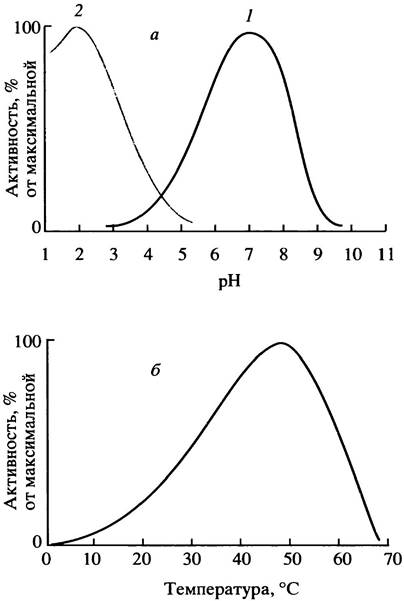
С другой стороны, имеет значение также ионизация субстрата. Иными словами, скорость катализируемой реакции обычно зависит от pH. Эта зависимость может быть различной, но обычно она характеризуется максимумом, расположенным в нейтральной области значений pH (типичная кривая показана на рис. 1.3, а). Случаются исключения. Так, пищеварительный фермент пепсин работает в сильно кислой среде желудка; рН-оптимум пепсина находится в области 2,0. Температура тоже влияет на активность ферментов. С ее ростом скорость катализируемых реакций сначала возрастает, примерно вдвое на каждые 10° С, а далее начинает падать из-за инактивации ферментов, поскольку они, как и большинство белков, неустойчивы к нагреванию. Типичная зависимость ферментативной активности от температуры приведена на рис. 1.3, б, однако на форму подобных кривых сильно влияют условий проведения эксперимента: чем короче время пребывания фермента при высокой температуре, тем меньше ее повреждающее действие. Хотя некоторые ферменты выдерживают нагревание, большинство из них инактивируется при температурах выше 50° С, а некоторые и при более низких. К этому следует добавить, что многие ферменты активны лишь в присутствии определенных ионов металлов, например, Мg2+, Zn2+, Fе2+ или Сu2+.
Рис. 1.3. Влияние pH (а) и температуры (б) на относительную активность ферментов: а - Кривая 1 с максимумом при физиологическом значении pH характерна для большинства ферментов, кривая 2 отвечает пепсину, функционирующему в сильнокислом содержимом желудка; б - резкое падение активности при высоких температурах связано с денатурацией фермента (есть и термостойкие ферменты). Положение максимума зависит от скорости нагревания
Активность ферментов изменяется также в присутствии ингибиторов. Вещества, принадлежащие к классу конкурентных ингибиторов, по строению подобны субстрату и конкурируют с ним за активный центр фермента. Неконкурентные ингибиторы уменьшают активность фермента, вступая с ним в разного рода специфические взаимодействия. Например, ион ртути образует ковалентное соединение с тиоловой группой, принимающей непосредственное участие в акте катализа. В других случаях ингибитор необратимо ацилирует фермент. Это происходит при действии аспирина на циклооксигеназу, которая участвует в биосинтезе простагландинов (подробности см. на с. 146).
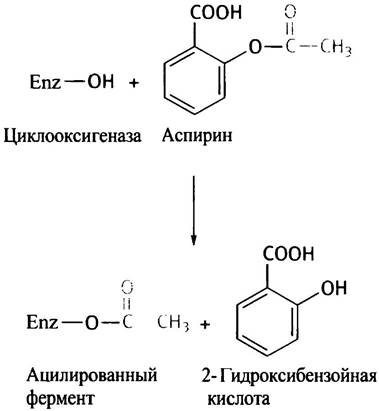
Ацилируемая группа здесь принадлежит остатку серина, который входит в состав активного центра циклооксигеназы.
Как расщепление пищи в клетках сопряжено с внутриклеточными реакциями, потребляющими энергию
Образование из низкомолекулярных предшественников таких биополимеров, как белки и ДНК, не может быть спонтанным, поскольку сопряжено со значительным увеличением свободной энергии. В термодинамическом контексте спонтанность означает отсутствие потребности во внешнем источнике свободной энергии. Внешним источником энергии является процесс окисления пищи. Химические превращения, протекающие с положительным изменением свободной энергии (синтетические, или «строительные» процессы) в совокупности называют анаболизмом, или анаболическими реакциями (анаболические стероиды, которыми злоупотребляют некоторые спортсмены, увеличивают массу тела - отсюда их название). Остальная часть метаболических превращений состоит из деструктивных процессов, протекающих с отрицательным изменением свободной энергии. Их называют катаболизмом, или катаболическими реакциями. Таким образом, метаболизм включает в себя анаболизм и катаболизм.
В результате катаболизма высвобождается свободная энергия, которая используется для приведения в действие энергопотребляющих анаболических реакций. Химическое превращение, происходящее с положительным изменением свободной энергии, требует ее притока извне, но значение ∆С, характеризующее процесс в целом, должно всегда быть отрицательным. Катаболические реакции, при которых энергия высвобождается, часто называют экзоэргоническими, а энергопотребляющие анаболические реакции — эндоэргоническими. Следует четко различать такие понятия, как химическая реакция и химическое превращение, т. е. процесс, происходящий в ходе ряда химических реакций. Так, соединение А в клетке реально может превратиться в соединение X, даже если для последнего характерна большая стандартная свободная энергия и его образование требует притока энергии извне. Однако это превращение протекает как последовательность отдельных химических реакций, каждая из которых имеет отрицательное значение ∆G. Обсудим кратко, как это может произойти.
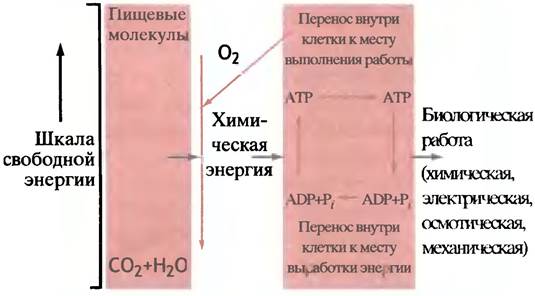
Такую совокупность реакций можно представить общей схемой, изображенной на рис. 1.4. В результате окисления пищи высвобождается энергия, которая используется для приведения в действие энергетически невыгодных процессов. Чтобы придать этой схеме глобальное значение, рассмотрим фотосинтез, при котором световая энергия используется для синтеза пищевых молекул (глюкозы) из СO2 и Н2O. Глюкоза превращается в организмах в другие пищевые молекулы, например, в жир. Хотя сборка крупных клеточных структур связана с уменьшением энтропии (т. е. невыгодна), окисление пищевых молекул сопровождается значительным увеличением энтропии (и потому выгодно). При этом для системы в целом (клетка и окружающая среда) изменение энтропии положительно, так что второй закон термодинамики выполняется в полной мере (согласно этому закону, общая энтропия Вселенной может только возрастать).
Рис. 1.4. Энергетический цикл жизни
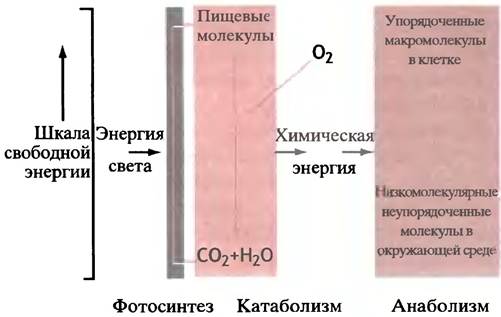
Здесь можно усмотреть аналогию с автомобилем, который поднимается в гору за счет сгорания бензина. Однако засунув пылающее ведро с горючим под капот, нужного эффекта не получишь, ибо должен существовать некий механизм для передачи выделяющейся энергии горения от ведра к колесам. Точно так же и клетка должна уметь сопрягать экзоэргонические и эндоэргонические реакции, чтобы разумным образом использовать энергию, которой иначе суждено бесполезно рассеяться в виде тепла.
Как это происходит? Использовать высвобождающееся тепло невозможно потому, что в физиологических условиях оно рассеивается. Энергия, высвобождающаяся при расщеплении пищевых веществ, используется в химических реакциях для совершения механической (мускульной) работы, в осмотических и электрофизиологических процессах. С другой стороны, для каждого потребляющего энергию процесса организму было бы слишком сложно создавать специфический преобразователь энергии. Вместо этого в клетках есть общий энергетический интермедиат, который принимает энергию от всех реакций окисления пищи и доставляет ее туда, где совершается работа и требуется энергия. Это более гибкий способ передачи энергии. Снова прибегнув к аналогии, уподобим его энергетике, опирающейся на электричество. Свободную энергию, выделяющуюся при сгорании угля, нефти, дерева или газа, превращают в электроэнергию, последняя же легко может быть передана туда, где она нужна, и использована так, как это требуется. (Впрочем, эта аналогия не вполне верна, ибо производство электроэнергии здесь опосредовано использованием тепла, чего нет в клетках.) Так мы приходим к мысли, что универсальный переносчик энергии должен иметь химическую природу. И потому закономерен вопрос: что же это за вещество?
Высокоэнергетический фосфат
Ответ на вопрос замечательно прост. Такое вещество универсально для всех форм земной жизни (строго говоря, исключения есть, но они столь экзотичны, что лишь подчеркивают универсальность).
Общую концепцию можно изложить предельно ясно: при распаде пищевых молекул неорганические фосфатные ионы превращаются в фосфорильные группы молекул так называемых макроэргических фосфатных органических соединений (что это значит, см. ниже). Эти молекулы в клетке транспортируются туда, где нужно совершить полезную работу. Такая работа совершается за счет энергии, запасенной в химических связях и выделяющейся при обратном превращении фосфорильных остатков в неорганические фосфатные ионы.
Тут крайне важно отметить, что ни в коем случае нельзя рассматривать обратное превращение фосфорильных остатков как обычный гидролиз, ибо при гидролизе вся выделяющаяся энергия превращается в бесполезное тепло. Рассмотрим упрошенный механизм, который позволяет «обуздать» энергию химических связей и использовать ее для совершения полезной работы.
Что такое «макроэргический фосфат»?
Внутриклеточные соединения, содержащие фосфорильные остатки, можно разделить на две группы. «Низкоэнергетические», при гидролизе которых до неорганического фосфата (Рi) отрицательное изменение свободной энергии ∆G°′ лежит в пределах 9-20 кДж • моль-1. И «высокоэнергетические», или макроэргические, у которых ∆G°′ превышает 30 кДж • моль-1. Понятие высокоэнергетический фосфат иногда используют для описания не самого соединения, а фосфатной связи. Однако это противоречит принятым в химии представлениям, согласно которым высокоэнергетическая связь - такая связь, расщепление которой требует больших затрат энергии. Поэтому мы будем для ясности говорить о высокоэнергетической фосфорильной группе, подразумевая, что присутствие такой группы придает высокую энергетичность молекуле в целом. С этой важной оговоркой именно высокоэнергетическую фосфорильную группу можно рассматривать как энергетическую «валюту» живой клетки. Концепцию иллюстрирует рис. 1.5, уточняющий рис. 1.4.
Рис. 1.5. Участие фосфатных групп в энергетическом хозяйстве клетки
Ⓡ-Фосфатная группа в молекулах, гидролиз которых приводит к отщеплению Рi и сопряжен с высвобождением более 30 кДж • моль-1
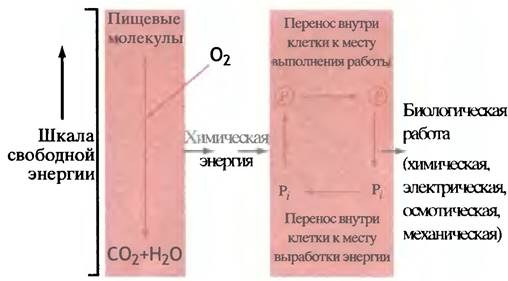
В чем главные структурные особенности высокоэнергетических фосфатов?
Фосфорная кислота Н3РO4 является гидроксипроизводным фосфора, содержащим в молекуле 3 протона.
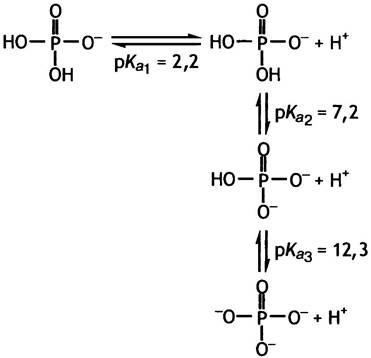
При физиологических значениях pH эта кислота в растворе представлена главным образом одно- и двухзарядными анионами, равновесную смесь которых биохимики обозначают сокращенно Рi, где индекс i (от англ. inorganic) указывает на неорганическую природу вещества. Смеси анионов (Рi) соответствует самый низкий уровень свободной энергии фосфата, который условно можно принять за исходный, или нулевой.
Степень ионизации фосфорной кислоты можно вычислить, исходя из значений трех констант ее диссоциации. Они могут быть представлены в виде параметров рКа (напомним, что под рКa понимают такое значение pH, при котором в диссоциированном состоянии находится половина соответствующих групп). Более подробные разъяснения рКаданы в приложении к этой главе. Его следует изучить, если вы еще не знакомы с такими понятиями, как диссоциация, буферные растворы и т. п. При физиологическом значении pH (скажем 7,4) группа с рКа 2,2 полностью диссоциирована, а группа с рКа 12,3 полностью находится в недиссоциированном состоянии. Группа с рКа 7,2 диссоциирована частично. Для вычисления степени диссоциации химических групп можно воспользоваться уравнением Гендерсона - Хассельбаха:
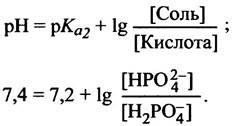
Таким образом,
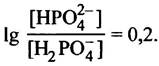
Откуда следует, что
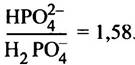
Неорганический фосфат, этерифицированный органическим спиртом, называют фосфоэфиром.
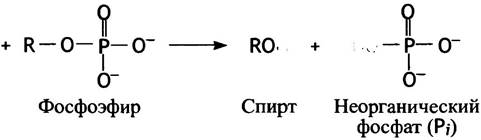
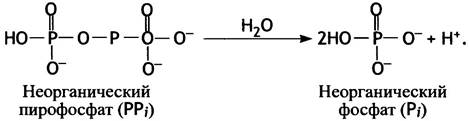
При гидролизе такого фосфоэфира величина ∆G°′ составляет около -12,5 кДж • моль-1. Это означает, что равновесие реакции сильно сдвинуто в сторону образования продуктов гидролиза и обратная реакция внутри клеток не происходит.
Точно так же, как фосфоэфиры образуются из неорганического фосфата и спиртов, из 2 молекул фосфата можно получить производное фосфорного ангидрида, которое называют пирофосфатом и обозначают РРi (пиро- означает огонь; пирофосфат может быть получен отщеплением воды от Рi при высокой температуре). Изменение свободной энергии при гидролизе этого вещества значительно больше (∆G°′ = -33,5 кДж • моль-1), чем для фосфоэфиров.
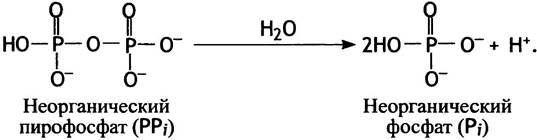
Столь значительное высвобождение свободной энергии при гидролизе пирофосфата обусловлено несколькими факторами.
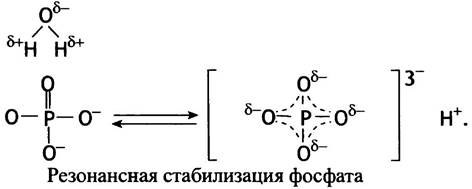
1. Структура пирофосфата дестабилизирована электростатическим отталкиванием двух отрицательно заряженных фосфорильных групп. То же самое можно представить иначе: при образовании пирофосфатной связи нужно сблизить одноименно заряженные остатки, преодолевая их отталкивание.
2. Продукты реакции гидролиза пирофосфатов (в простейшем случае 2 Рi) резонансно стабилизированы, т. е. имеют большее число возможных резонансных структур, чем структура пирофосфата. Это происходит потому, что фосфорильные группы в пирофосфатах конкурируют за электроны мостиковых атомов кислорода. Наличие таких электронов необходимо для образования резонансных структур. В результате конкуренции то одна, то другая фосфорильная группа в РРi может резонировать, тогда как два Рiмогут резонировать одновременно. Кроме того, в неорганическом фосфате все Р-О связи усреднены по свойствам, поэтому протон не связан с каким-то определенным атомом кислорода, что автоматически приводит к увеличению энтропии. На приведенной ниже схеме d- обозначает частичный отрицательный заряд (d+ означал бы частичный положительный заряд).
Резонансная стабилизация фосфата
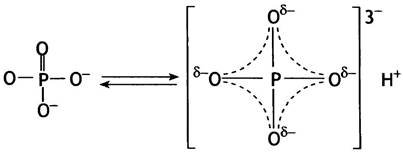
Все эти факторы способствуют протеканию реакции гидролиза РРi, равновесие которой сдвинуто в сторону образования Рi. Эти рассуждения справедливы по отношению не только к самому пирофосфату, но и к любому соединению, содержащему фосфорноангидридную группу, о которых речь пойдет далее. Отметим, что к обычным сложным эфирам фосфата представленные выше рассуждения неприложимы; именно поэтому при их гидролизе высвобождается гораздо меньше энергии.
Пирофосфаты хотя и главные, но не единственные биологические макроэргические производные фосфорной кислоты. Известны еще три типа таких соединений.
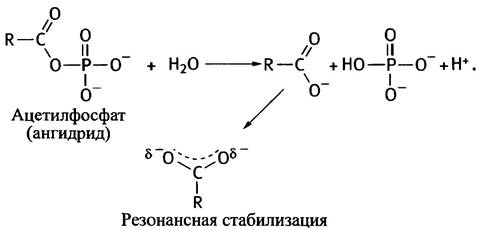
К первому относятся смешанные ангидриды фосфорной и карбоновых кислот, гидролиз которых характеризуется величиной ∆G°′ около -49 кДж • моль-1. Столь большое значение ∆G°′ объясняется резонансной стабилизацией обоих продуктов гидролиза: Рi и карбоксилатного аниона.
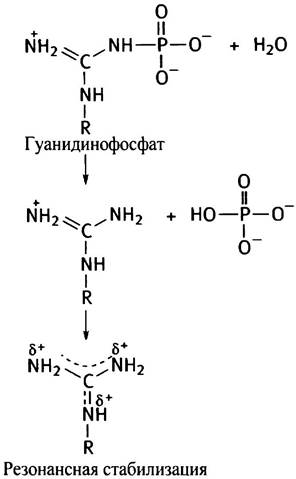
Ко второму типу соединений относятся гуанидино- фосфаты, у которых величина ∆G°′ благодаря резонансной стабилизации обоих продуктов гидролиза составляет около -43 кДж • моль-1.
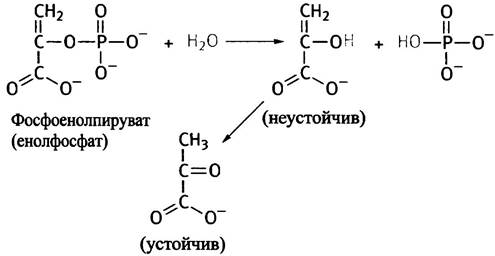
К третьему типу соединений принадлежат енолфосфаты, имеющие совсем иное строение. Рассмотрим одно из таких соединений - фосфоенолпируват. На первый взгляд в нем трудно угадать макроэргическое соединение, однако при гидролитическом отщеплении фосфатной группы образуется енольная форма пирувата, которая спонтанно таутомеризуется в кетоформу. При этом равновесие сдвинуто в сторону образования последней, благодаря чему суммарный процесс превращения фосфоенолпирувата в пируват и фосфат протекает с выделением энергии ∆G°′ = -61,9 кДж • моль-1.
В последующих главах этой книги вы еще не раз будете встречаться с различными производными фосфорной кислоты и обсуждать свойственные им значения ∆G°′, однако сейчас мы переходим к наиболее общему и важному в этой проблеме.
До сих пор мы говорили о присутствии фосфорильной группы в высокоэнергетических фосфатах. На рис. 1.5 показано, что неорганический фосфат преобразуется в клетке в «высокоэнергетическую фосфорильную группу», которая далее транспортируется к месту потребления энергии для совершения работы. Ясно, что такая группа не существует сама по себе, а является частью молекулы и перемещается вместе с ней. Возникает нижеследующий вопрос...
Что в клетке служит переносчиком фосфорильной группы?
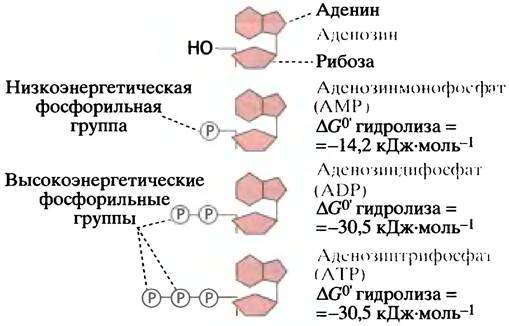
Главным переносчиком фосфорильной группы является аденозинмонофосфат - АМР, структура которого изображена на рис. 1.6. АМР, несущий еще одну фосфорильную группу, называют аденозиндифосфатом -ADP. Если фосфорильных групп три, то соединение называется аденозинтрифосфатом - АТР. Две концевые фосфорильные группы в АТР связаны между собой точно такой же фосфоангидридной связью, как и в пирофосфате. И точно так же, как пирофосфат, это соединение гидролизуется с выделением большой свободной энергии. Заметим, что этим АТР принципиально отличается от АМР, который в цикле переноса энергии выступает лишь как переносчик пирофосфатных остатков.
На рис. 1.6 показано, что гидролиз каждой из двух концевых фосфатных групп в молекуле АТР характеризуется величиной ∆G°′, равной около -30,5 кДж • моль-1: следовательно, обе они могут рассматриваться как высокоэнергетические.
Рис. 1.6 Аденозин и его фосфорилированные производные. Аденозин (изображен схематически) - это нуклеозид, состоящий из аденина и рибозы
Окисление пищи сопряжено с превращением ADP в АТР:
![]()
В каждый данный момент любая клетка содержит небольшое количество АТР, которого ей хватает лишь на короткое время. Ни АТР, ни ADP, ни АМР, молекулы которых заряжены, не могут спонтанно диффундировать через клеточную мембрану (см. главу 3). Поэтому каждая клетка вынуждена сама вырабатывать АТР, поддерживая его быстрый кругооборот, включающий гидролиз АТР до ADP и фосфата и последующий ресинтез АТР из этих соединений. С учетом изложенного, мы можем модифицировать схему, представленную на рис. 1.5 (рис. 1.7). Яд цианид в основном блокирует внутриклеточный ресинтез АТР, и смерть наступает почти мгновенно из-за того, что энергопотребляющие процессы останавливаются, как только резерв АТР будет исчерпан.
Рис. 1.7. Участие АТР в энергетическом хозяйстве клетки. На универсальности этого рисунка не сказываются отдельные случаи, когда с выполнением работы сопряжен гидролиз АТР до АМР
Как АТР подвергается химическим превращениям?
Предположим, клетке требуется синтезировать соединение Х-Y из двух компонентов, Х-ОН и Y-H, причем для этого процесса значение ∆G°′ равно 12,5 кДж • моль-1. Прямая реакция Х-ОН + Y-H —> X-Y + Н2O не пойдет, поскольку она эндоэргонична и равновесие сильно смещено влево. Ниже показано, как решается эта проблема путем сопряжения синтеза Х-Y с гидролизом АТР. Сопряженными будем называть те реакции, когда продукт одной из них является исходным веществом для другой. При этом речь идет только о системе реакций в чисто химическом смысле. Все внутриклеточные реакции с участием АТР катализируются ферментами. Тот факт, что гидролиз каждой из двух фосфорноангидридных групп сильно экзоэргоничен, не означает, что АТР является нестабильным или высокореакционноспособным соединением (в виде белого порошка его можно хранить неопределенно долго в обычном холодильнике).
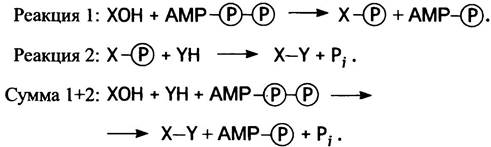
Результирующее значение ∆G°′ здесь должно быть арифметической суммой величин ∆G°′, соответствующих реакциям 1 и 2. Поскольку ∆G°′ для реакции Х-ОН + YH —> X-Y + Н2O составляет 12,5 кДж • моль-1, а для гидролиза АТР (АТР + Н2O —> ADP + Рi) ∆G°′ = -30,5 кДж • моль-1, результирующее значение ∆G°′ = -18,0 кДж • моль-1. Это означает, что суммарный процесс сильно экзоэргоничен и, следовательно, протекает вплоть до практически полного завершения (константа его равновесия ~103, т. е. в равновесных условиях на 99,9% смещено в сторону Х-Y; см. табл. 1.1). Заметим, что здесь имеет место не прямой гидролиз АТР до ADP и фосфата. Фосфатная группа сначала переносится на молекулу одного из реагирующих компонентов и лишь потом отщепляется в свободном виде.
В клетке реальное значение ∆G°′ для любых процессов гидролиза АТР до ADP и фосфата гораздо больше стандартной величины ∆G°′, поскольку концентрации всех участвующих в реакции веществ существенно меньше 1М. Если истинные концентрации известны, свободную энергию можно вычислить с помощью уравнения:
![]()
где R - газовая постоянная (8,315 Дж • моль-1 • К-1) а T - температура в градусах Кельвина (298 К = 25° С). Хотя концентрации АТР, ADP и Рi в различных клетках неодинаковы, в среднем они близки к 10-3 М для АТP и Рi и 10-4М для ADP. Для простоты предположим, что все эти концентрации равны 1 мМ. Подставляя это значение в уравнение:
![]()
получаем:
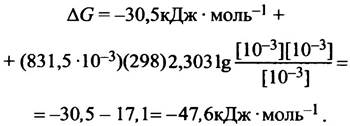
Описанная выше последовательность реакций характерна для многих биосинтетических процессов, сопряженных с расщеплением АТР. Однако при биосинтезе таких макромолекул, как белки и нуклеиновые кислоты, природа использует более эффективный энергетический трюк: от АТР отщепляется не одна, а две фосфатные группы. Благодаря чему отрицательное изменение ∆G столь велико, что эти процессы протекают до полного завершения и совершенно необратимы. Как это достигается, мы покажем, опять-таки, на примере той же реакции Х-ОН + YH —> X-Y + Н2О.
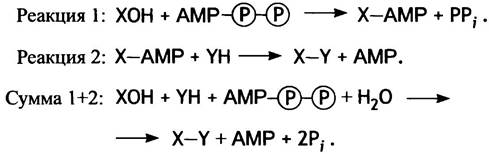
Предположим, как и ранее, что для реакции Х-ОН + YH —> X-Y + Н2О изменение ∆G°′ составляет 12,5 кДж • моль-1, ∆G°′ для реакции АТР + Н2О —> АМР + РРi∆G°′ равно -32,2 кДж • моль-1, а для гидролиза PPi∆G°′ равно -33,5 кДж • моль-1.
Реакции АТР + Н2O —> АМР + 2Рi соответствует ∆G°' = -65,7 кДж • моль-1, а итоговому синтезу X-Y, протекающему сопряженно с этой реакцией, будет отвечать весьма значительная отрицательная величина ∆G°' = -65,7 - 12,5 = -53,2 кДж • моль-1.
Эффективность суммарной реакции зависит от того, насколько быстро в клетке расщепляется неорганический пирофосфат РРi. Его гидролиз катализирует специальный фермент пирофосфатаза, который можно назвать движущей силой важнейших процессов биосинтеза.
Как с помощью АТР совершаются другие виды работ?
Энергия, высвобождающаяся при расщеплении АТР, помимо участия в химических реакциях обеспечивает мышечное сокращение, генерацию электрических сигналов, а также перекачку ионов и молекул против градиентов их концентраций. Механизмы всех этих процессов, которые подробнее будут рассмотрены позже, принципиально сходны в том, что для их осуществления используется свободная энергия, освобождающаяся при расщеплении АТР.
Замечание о соотношении между АМР, ADP и АТР
Выше было сказано о том, что во многих случаях ATP-зависимый синтез химических соединений в клетке приводит к образованию не ADP + Рi, а АМР + РРi. В то же время АМР, в отличие от ADP, не превращается в АТР при окислении пищевых веществ (см. рис. 1.6). Какова же судьба образующегося АМР? Ответ можно найти в обратимой реакции переноса фосфатного остатка с АТР на АМР: АМР + АТР <-> 2 ADP. Ее катализирует фермент AMP-киназа. Киназами называют ферменты, осуществляющие перенос фосфатною остатка с АТР на молекулу акцептора. Термин AMP-киназа означает, что акцептором фосфата является АМР (поскольку другое название АМР - адениловая кислота; этот же фермент иногда называют аденилаткиназой). Заметим, что фосфатный остаток при этом переносится непосредственно от одной молекулы к другой, а изменение свободной энергии относительно невелико. Далее вы убедитесь, что такого рода «перетасовка» энергетических ресурсов широко распространена. Образующийся в данном случае ADP может быть вовлечен в синтез АТР, сопряженный с окислением пищевых веществ.
Детальному рассмотрению механизмов, посредством которых при окислении пищи из ADP и Рi образуется АТР, будет уделено особое внимание в главах, посвященных метаболизму. Здесь мы лишь в самом общем виде затронули вопросы, касающиеся использования энергии АТР для совершения полезной работы. Конкретные механизмы утилизации энергии АТР будут подробно рассмотрены в следующих главах.
А сейчас мы несколько изменим тему, хотя и продолжим анализ изменений свободной энергии в химических процессах.
Слабые связи и изменения свободной энергии
До сих пор речь шла о явлениях, связанных с образованием и расщеплением ковалентных связей. Теперь пора перейти к рассмотрению нековалентных связей, называемых также вторичными, или слабыми (два последних эпитета не отражают их значимости для жизни).
В реакциях, катализируемых ферментами, происходит перераспределение связей между атомами, однако суммарное число связей при этом сохраняется. Когда два атома соединяются посредством химической связи, выделяется определенное количество энергии:
X + X —> X - X + энергия, где X - свободный атом.
Два таких атома соединяются связью, которая ранее не существовала. Мы не рассматриваем здесь обычные химические реакции, в ходе которых число ковалентных связей не увеличивается.
Чтобы обратить рассмотренное превращение и разорвать химическую связь, нужно затратить столько же энергии, сколько выделилось при образовании связи:
X - X + энергия —> X + X.
Образование обычной ковалентной связи происходит с выделением значительной энергии; следовательно, значительная энергия должна быть затрачена и на ее расщепление. Такого рода расщепления в биохимических системах не происходят, что можно проиллюстрировать на примере кислорода, который существует в виде молекул O2. Их образованию из атомарного кислорода отвечает большое значение ∆G°′, около -460 кДж • моль-1, поэтому равновесное содержание атомарного кислорода исчезающе мало.
В обычных химических реакциях расщепление одной химической связи сопровождается одновременным образованием другой ковалентной связи через переходное состояние. При этом суммарное изменение энергии оказывается гораздо меньше, чем необходимо для расщепления ковалентных связей. Однако нековалентные слабые связи между отдельными атомами или группами атомов в растворе непрерывно возникают и исчезают. Они достаточно прочны, чтобы участвовать в формировании структуры вещества, но не настолько устойчивы, чтобы исключить разрушение этой структуры. Иными словами, такие связи определяют гибкость структуры молекулы. Их основополагающее значение выявится при рассмотрении мембран, белков и ДНК. В действительности эти связи слабы и вторичны не по их значению, а лишь по энергии их образования (∆G°′ = -4 ÷ 30 кДж • моль-1), которая достаточно мала для того, чтобы они легко разрывались. Известны три типа слабых связей: ионные, водородные и ван-дер-ваальсово притяжение.
Ионные связи возникают как следствие электростатического притяжения между достаточно близко расположенными группами, несущими постоянные по величине заряды. Типичным примером таких связей может служить взаимодействие между отрицательно заряженной карбоксилатной и положительно заряженной аммонийной группами:
-СОO- .......................... +NH3-.
Связи также возникают вследствие электростатического притяжения между атомами полярных молекул, однако здесь речь идет о гораздо менее выраженном
разделении зарядов, чем в случае ионных связей. А именно - об электрическом дипольном моменте. Рассмотрим в качестве примера распределение зарядов в молекуле воды.
Атом кислорода в молекуле воды электроотрицателен. Это означает, что он оттягивает на себя электроны, образующие связь более эффективно, чем атомы водорода. В результате в молекуле воды атом кислорода приобретает частичный отрицательный заряд, а атомы водорода - соответствующие частичные положительные заряды. Это приводит к возникновению между атомами О и Н соседних молекул воды слабых притяжений, называемых водородными связями. Каждая молекула воды может быть связана с 4 другими молекулами, поскольку атом кислорода участвует в образовании 2 водородных связей, а каждый атом водорода - в одной. Не только в воде, но и в других молекулах атомы водорода участвуют в образовании водородных связей, если они ковалентно присоединены к электроотрицательным атомам. В биохимических структурах роль последних чаще всего выполняют атомы кислорода и азота. Кроме водорода в образовании водородной связи участвует и его партнер - электроотрицательный акцепторный атом, обычно также кислород или азот.
Здесь показаны наиболее часто встречающиеся типы водородных связей.
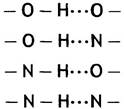
Водородная связь характеризуется определенной направленностью. Ее прочность максимальна, когда все вовлеченные в нее атомы расположены на одной прямой.
О ван-дер-ваальсовых силах говорят при описании слабых взаимодействий между тесно сближенными атомами, когда флуктуации электронной плотности в таких атомах индуцируют образование диполей. Эти силы неспецифичны и являются единственным типом слабых связей, возникающих между неполярными молекулами, которые неспособны образовать ионные и водородные связи. Ван-дер-ваальсовы силы действуют между любыми двумя атомами, если они сближены настолько, что их электронные оболочки почти соприкасаются. При этом притяжение обратно пропорционально расстоянию между атомами в шестой степени. Отсюда понятна необходимость тесного сближения атомов. Однако, если атомы заставить сблизиться еще больше, их электронные оболочки начнут перекрываться, что приведет к возникновению сил отталкивания. Таким образом, ван-дер-ваальсовы силы эффективны лишь в узком диапазоне межатомных расстояний.
Все три типа слабых связей объединяет то, что они возникают как короткодействующее притяжение. Различаются же они своей длиной и энергией образования: ∆G°′ для ионных связей составляет около 20 кДж • моль-1, для водородных - 12 ÷ 19 кДж • моль-1, а для ван-дер-ваальсовых - 4 ÷ 8 кДж • моль-1. Для сравнения: энергия образования ковалентной связи составляет сотни кДж • моль-1.
Значение слабых связей состоит в том, что именно они ответственны за внутри- и межмолекулярную ассоциацию молекул. Здесь необходимо вспомнить о факторе, который также способствует ассоциации, хотя его и нельзя отнести к подлинному связыванию: речь идет о так называемом гидрофобном связывании, или гидрофобном взаимодействии.
Полярные вещества вроде сахара растворимы в воде, потому что их молекулы хотя и нарушают водородные связи между молекулами воды, но и сами образуют с ними такие связи, что в целом оказывается энергетически предпочтительным. Соли, например, NаСl, растворимы в воде потому, что вокруг ионов Na+ и Сl- возникают гидратные оболочки, которые взаимодействуют с ионами сильнее, чем эти ионы между собой. В результате ионы расходятся, а это, в свою очередь, приводит к значительному росту энтропии при переходе от кристалла к раствору. Что произойдет, если попытаться растворить в воде неполярное вещество, скажем оливковое масло? Его молекулы не могут образовать водородные связи с окружающими молекулами воды. Это означает, что у пограничных с маслом молекул воды некоторая доля потенциально способных к связыванию атомов останется без партнеров. Чтобы уменьшить число подобных атомов, молекулы воды перестраиваются таким образом, что приобретают упорядоченность большую, чем в обычной воде (энтропия уменьшается). Это препятствует растворению в воде масла и других неполярных веществ - и одновременно вызывает слипание отдельных неполярных молекул, поскольку при этом уменьшается совокупная площадь их контакта с молекулами воды. Внешне это выглядит как подталкивание неполярных молекул друг к другу. Силы, действующие при этом, принято называть гидрофобными. Гидрофобные группы присутствуют
и в белках, и в ДНК, и в других внутриклеточных молекулах и потому играют важную роль в формировании их структуры. Просто удивительно, сколь многое в живых клетках определяется необходимостью «спрятать» гидрофобные группы от воды!
Что вызывает образование и разрыв слабых связей?
Образование слабых связей происходит с уменьшением свободной энергии системы и не требует преодоления сколько-нибудь заметного активационного барьера. Поэтому такие связи возникают между подходящими и притом достаточно сближенными атомами совершенно спонтанно, без участия ферментов. Мы уже упоминали о соотношении, связывающем константу равновесия реакции с величиной ∆G°′ (результаты соответствующих вычислений представлены в табл. 1.1). Из них следует, что даже в том случае, когда при образовании слабой связи ∆G°′ составляет всего -5,7 кДж • моль-1, соответствующее равновесие реакций сильно смещено в сторону связывания. Если же ∆G°′ = -11,4 кДж • моль-1, степень связывания достигает 99%. Отсюда видно, насколько образование слабых связей чувствительно к величине изменения свободной энергии.
Нельзя забывать, что равновесие - динамический процесс: отдельные связи постоянно разрываются и образуются вновь. Что вызывает разрыв связи? Для этого достаточно передать связанным атомам такое же количество энергии, какое ранее высвободилось при образовании данной связи. Эта энергия черпается из кинетической энергии соседних молекул, соударяющихся со связанными атомами благодаря тепловому движению. Даже при физиологических значениях температуры кинетической энергии наиболее быстрых молекул вполне хватает, чтобы разорвать слабую связь.
Если слабые связи так легко рвутся, каково их значение в биохимических системах?
Возьмем, например, ван-дер-ваальсово притяжение. Отвечающая ему свободная энергия едва превышает тепловую энергию молекул в растворе, и соответствующие связи постоянно образуются и разрываются. Неужели столь эфемерное взаимодействие может иметь какое- либо значение? Хотя и в меньшей степени, но это относится и к другим типам слабых связей.
Как ни парадоксально, именно слабость таких связей определяет их центральную роль в явлениях жизни. Их главная особенность состоит в исключительной
специфичности. Взаимное узнавание биологических молекул происходит с такой исключительной точностью, что связываются лишь те из них, которые отобраны для этого эволюцией.
Вот несколько примеров. Фермент избирательно ускоряет только одну определенную реакцию: прежде всего потому, что его активный центр связывает только один или немногие из потенциально способных к образованию такой связи субстратов. Гормон оказывает строго определенное действие, поскольку он связывается не с любым, а только с предназначенным для него белком- рецептором. Функционирование генов зависит от специальных белков, которые распознаются и связываются с определенными участками ДНК. Антитело образует комплекс только с соответствующим ему антигеном. Этот список необъятен, и по мере изучения биохимии вы будете встречать все новые и новые примеры специфичности. Как же обеспечивается столь высокая избирательность при межмолекулярных взаимодействиях? Именно благодаря слабым связям. При этом важно нижеследующее.
1. Объединение молекул в устойчивый нековалентный комплекс возможно лишь при одновременном образовании не одной, а многих слабых связей.
2. Слабые связи требуют тесного сближения взаимодействующих атомов, а водородные связи, кроме того, их определенной взаимной ориентации.
Поэтому связываются только те молекулы-партнеры, которые по своей структуре комплементарны друг другу. Это правило соблюдается настолько безукоризненно, что иногда достаточно поменять всего один атом в молекуле субстрата или активном центре фермента, чтобы они перестали взаимодействовать.
Наряду со специфичностью важнейшей особенностью взаимодействий в биологических системах является их транзиторный характер. Связывание в активном центре фермента не может быть очень прочным. И функционирование гена, и контроль за ним также предполагают легкую обратимость ответственных за это взаимодействий. Это жестко ограничивает диапазон допустимых для таких взаимодействий отрицательных значений ∆G°′ (т. е. констант равновесия): они должны быть достаточно велики, чтобы слабые связи легко образовывались, и вместе с тем достаточно малы, чтобы эти связи легко нарушались, обеспечивая в целом необходимую степень обратимости взаимодействия. В случае же взаимодействия типа «антиген - антитело» большое число слабых связей приводит к практически необратимому связыванию. Итак, слабые связи позволяют точно дозировать межмолекулярные взаимодействия и лежат в основе биологической избирательности. Если таких связей предусмотрено немного, то взаимодействие вполне обратимо, в противном же случае образуются прочные меж- или внутримолекулярные взаимодействия. Роль слабых связей в биохимических системах станет выглядеть еще более значимой при дальнейшем знакомстве с мембранами, структурой белков, строением и функционированием генов.
Таким образом, все изложенное выше можно рассматривать как предисловие к двум последующим главам о структуре белков и биологических мембранах. Обе эти темы необходимы для понимания метаболизма, которому посвящена первая половина книги.
Буферные растворы и значения рКа
Биохимику совершенно необходимо понимать, что такое буферные растворы и что такое рКа. Удобно выделить эту тему в отдельный раздел, дабы она не затерялась в основном тексте.
В живых клетках pH поддерживается в пределах 7,2 -7,4. Лишь в некоторых особых ситуациях pH выходит за эти пределы, например, в полости желудка, где секретируется НСl, или в лизосомах, куда специально закачиваются протоны. Постоянство pH соблюдается несмотря на то, что при метаболических процессах происходит, скажем, образование молочной и ацетоуксусной кислот или имеет место характерное для крови превращение СO2 в Н2СО3.
В значительной мере постоянство pH обеспечивается буферным действием слабых кислот. Термин слабая означает, что в данных условиях такая кислота лишь частично диссоциирована.
Карбоновая кислота диссоциирует, высвобождая протон:
R-COOH <-> R-СОO- + Н+
Донор и акцептор протона в этом и подобных ему уравнениях называют сопряженными кислотой и основанием. В общем виде уравнение диссоциации кислот можно записать так:
НА <-> Н+ + А-
Кислоты различаются по склонности к диссоциации. Говоря о том, что одна кислота более сильная, а другая - более слабая, имеют в виду, что первая из них диссоциирует легче. Так, в растворе 0,1 М муравьиной кислоты (НСООН) pH меньше, чем в растворе уксусной кислоты (СН3СООН): последняя является более слабой. Способность кислоты к диссоциации можно количественно выразить константой диссоциации Ка; чем она больше, тем в большей степени кислота диссоциирует, т. е. тем она сильнее:
![]()
Для уксусной кислоты Ка = 1,74 • 10-5. Вместо константы диссоциации биохимики гораздо чаще используют родственный параметр рКа, который связан с ней уравнением: pKa = -lg Ka
Таким образом, рКа уксусной кислоты равно 4,76, а муравьиной - 3,75. Эти значения представляют собой pH, при котором кислота наполовину диссоциирована. С увеличением pH степень диссоциации кислоты возрастает, а с уменьшением pH падает. О влиянии концентрации Н+-ионов на положение равновесия реакции диссоциации НА <-> Н+ + А-можно судить по данным, представленным на рис. 1.8.
Рис. 1.8. Влияние pH на ионизацию -СООН и -NH2 групп (данные любезно предоставлены R. Rogers)
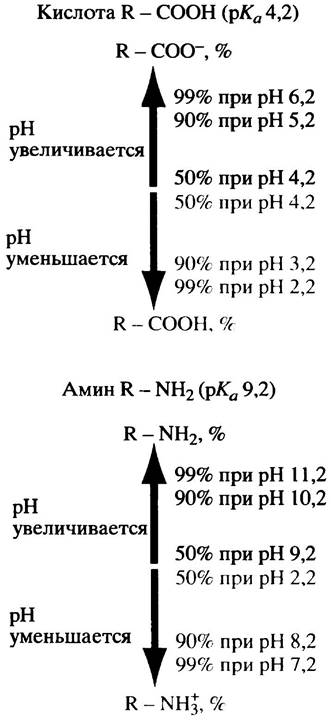
Амины тоже имеют свои рКа, поскольку их протонированная (аммонийная) форма может диссоциировать с отщеплением протона и, соответственно, рассматриваться как кислота.
R-NH3+ <-> R-NН2 + Н+
В этом случае степень диссоциации уменьшается с ростом pH, тогда как у карбоновых кислот она возрастает. Увеличение концентрации Н+ приводит к увеличению протонирования как карбоновых кислот, так и аминных оснований, с той разницей, что в первом случае количество ионизированных форм уменьшается, а во втором - увеличивается (см. рис. 1.8).
Как связаны значения рКа и буферные растворы
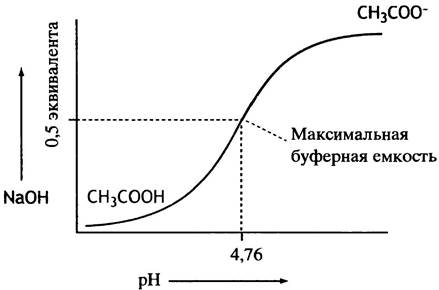
Если понемногу добавлять раствор 0,1 М NаОН к раствору 0,1 М уксусной кислоты и после каждой такой добавки измерять pH, то можно построить график, приведенный на рис. 1.9. Такого рода эксперимент называется титрованием. В начале титрования добавляемые ионы ОН нейтрализуют присутствующие в растворе ионы Н+ (уксусная кислота слегка диссоциирована), а pH при этом увеличивается быстро. Однако, по мере того как pH приближается к рКа уксусной кислоты, в ответ на каждую добавку щелочи все большая доля уксусной кислоты диссоциирует с образованием ацетата и водородных ионов. Последние все в большей степени нейтрализуют добавляемые ионы гидроксила, так что в результате изменения pH становятся все меньше. С учетом того, что ацетат натрия полностью диссоциирован, реакцию можно описать уравнением:
СН3СООН + ОН- —> СН3СОО- + Н2O.
Рис. 1.9. Кривая титрования уксусной кислоты (рKa 4,76)
Точно так же по другую сторону рКа по шкале pH добавление в среду ионов Н+ вызывает относительно небольшие изменения pH благодаря следующей реакции:
СН3СОO- + Н+ —> СН3СООН.
В этом и заключается буферный эффект уксусной кислоты. Он максимален вблизи рКа, где уксусная кислота и ацетат присутствуют в эквимолярных количествах. Биохимики пользуются таким эмпирическим правилом: буфер эффективен в интервале единицы pH, центр которого приходится на величину рКа. Для вычисления pH раствора, содержащего смесь сопряженной пары кислота-основание, можно использовать уравнение Гендерсона-Хассельбаха:
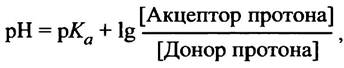
которое обычно записывают в виде:
![]()
Оно применимо для любой смеси уксусной кислоты и ацетата, что позволяет подобрать ацетатный буферный раствор с нужным значением pH. Для раствора, содержащего 0,1 М уксусной кислоты и 0,1 М ацетата натрия,
![]()
Если же раствор содержит 0,1 М уксусной кислоты и 0,2 М ацетата натрия, то
![]()
Предположим, что к раствору 0,1 М уксусной кислоты и 0,1М ацетата натрия добавлена щелочь до концентрации 0,05 М. Тогда половина уксусной кислоты будет превращена в ацетат. После этого величина pH составит:
![]()
Смесь ацетат-уксусная кислота не обладает буферным действием при физиологических значениях pH. Однако в организмах есть вещества, рКа которых позволяет им служить эффективными буферами при рН~7. Важнейшими среди таких веществ являются фосфатный ион и его производные. При диссоциации фосфорной кислоты может отщепляться 3 протона, причем отщеплению второго (Н2РO4- <-> НРО2-4 + Н+) соответствует рКа 6,86. Поэтому фосфат служит превосходным внутриклеточным буфером.
Буфером могут служить также имидазольные радикалы в остатках аминокислоты гистидина, входящей в состав белков. Протонированию этих радикалов отвечает рКа ~6.
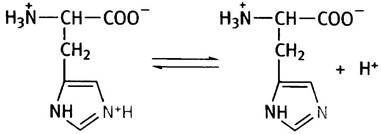
Вы можете наблюдать роль буфера, поставив простейший опыт. Наполните пробирку дистиллированной водой, доведя ее pH до 7 с помощью разбавленной щелочи (дистиллированная вода обычно слегка кислая из-за растворенного в ней СO2). Если к воде прибавить несколько капель разбавленной соляной кислоты, ее pH резко уменьшится. Проделайте то же с децимолярным раствором фосфата натрия: pH раствора почти не изменится. Вот это и есть буферное действие фосфата.
Таким образом, буферное действие веществ с рKa ~7 предохраняет клетки и межтканевые жидкости от значительных изменений pH.
Вопросы к главе 1
1. Представьте себе мужчину весом 70 кг, питание которого позволяет удовлетворить его энергетические затраты - 10 000 кДж в день. Допустим, что свободная энергия, которую он получает с пищей, используется для образования АТР из ADP и Рi с эффективностью 50%. В клетке ∆G превращения ADP + Рi приблизительно равно 55 кДж • моль-1. Вычислите, сколько весит АТР, синтезируемый этим человеком за один день (количество выразить в граммах динатриевой соли АТР с молекулярной массой 551).
2. Стандартная величина изменения свободной энергии гидролиза АТР до ADP и фосфата составляет -0,5 кДж • моль-1. Объясните, почему в предыдущем вопросе значение 55 кДж • моль-1 считается количеством свободной энергии, необходимой для синтеза 1 моля АТР из ADP и Pi в клетке.
3. Почему ∆G°' гидролиза АТР в ADP + Рi и ÂDP в АМР + Рi составляет -30,5 кДж • моль-1, тогда как для гидролиза АМР до аденозина и Рi величина ∆G°' = -14,2 кДж • моль-1? В чем причина такого большого различия?
4. Какую информацию можно извлечь из кривой температурной зависимости ферментативной активности?
5. Объясните, почему:
а) бензол практически нерастворим в воде;
б) полярные молекулы глюкозы растворимы в воде;
в) NaCl растворим в воде.
6. Катаболизм пищи в клетке устроен так, что обеспечивает превращение ADP в АТР, но не может непосредственно превратить АМР в АТР. Между тем многие ферментные системы превращают АТР в АМР. Как организм справляется с накоплением АМР?
7. Фермент катализирует синтез соединения X-Y из ХОН и YH, сопряженный с расщеплением АТР до АМР и неорганического пирофосфата. Стандартное изменение свободной энергии при реакции ХОН + YH —> X-Y + Н2O составляет 10 кДж • моль-1. Вычислите ∆G°' ферментативной реакции в клетке, а также при использовании очищенного фермента. Объясните полученные результаты. (В расчетах рекомендуем использовать для гидролиза АТР до АМР и РРi величину ∆G°' = -33,2 кДж • моль-1, а для гидролиза РРi∆G°' = -33,4 кДж • моль-1.)
8. Какие типы слабых связей используются в биологических системах и каковы их энергетические характеристики? Почему для образования этих связей не нужен ферментативный катализ? Каково значение слабых связей?
9. Диссоциация фосфорной кислоты описывается тремя рКа: 2,2; 7,2; 12,3.
а) Какая из ионных форм доминирует при pH 0, pH 4, pH 9 и pH 14?
б) Вычислите pH водного раствора эквимолярной смеси NaH2PO4 и Na2HPO4.
в) Какую аминокислоту из числа природных вы выбрали бы в качестве буфера для поддержания физиологического значения pH?
10. Окисление глюкозы до СO2 и воды протекает с выделением очень большого количества энергии. Как объяснить тот факт, что глюкоза на воздухе вполне устойчива к окислению?
11. Как ферменты катализируют химические реакции?