Биохимия - Химические реакции в живой клетке Том 3 - Д. Мецлер 1980
Свет в биологии
Поглощение света веществом
Электронные спектры
В биохимии широко используется спектроскопия в ультрафиолетовой (УФ) и видимой областях. Видимый свет занимает на шкале электромагнитных волн диапазон от ~ 12 000 см-1 (800 нм) до 25 000 см-1 (400 нм). Далее идет ультрафиолетовая область; максимальная частота, еще использующаяся в обычных спектрофотометрах, составляет ~55 000 см-1 (180 нм). Значения энергии, соответствующие видимому и ультрафиолетовому свету, лежат в интервале от ~ 140 до ~660 кДж∙моль-1. Отметим, что второе значение больше энергии любой связи, за исключением наиболее прочных двойных и тройных связей (табл. 3-6). Отсюда понятна способность УФ-излучения инициировать фотохимические реакции. Даже красный свет с его относительно низкой энергией используется растениями в фотосинтезе и несет достаточно энергии на 1 Эйнштейн, чтобы обеспечить генерацию АТР, восстановление NADP+ и ряд других фотохимических процессов. Хотя энергия поглощаемого при электронных переходах света довольно велика, геометрия молекул в возбужденном и основном состояниях, как правило, различается лишь весьма незначительно. В общем случае возрастает амплитуда колебаний и размеры молекулы немного увеличиваются в одном или нескольких направлениях. Ценность спектрофотометрических методов для биохимических исследований частично обусловлена высокой чувствительностью электронных энергетических уровней молекул к их непосредственному окружению. Точность и высокая чувствительность спектрофотометров в немалой степени расширяет возможности электронной спектроскопии. Повсеместно применяются и родственные методы — круговой дихроизм и флуоресценция. Наличие в белках, нуклеиновых кислотах, коферментах и многих других биохимических соединениях интенсивно поглощающих хромофоров еще более способствует популярности всех этих методов.
а. Форма полос поглощения
Электронные полосы поглощения обычно довольно широки: их ширина, измеряемая на уровне полувысоты, составляет 3000—4000 см-1. Связано это главным образом с тем, что электронное возбуждение сопровождается переходом молекулы на более высокие колебательные и вращательные подуровни. Вклад в уширение полос вносит также неоднородность окружения молекул в растворе. Форма полос поглощения в некотором приближении определяется принципом Франка—Кондона.
Поскольку частота света, поглощаемого при электронных переходах, составляет ~1015—1016 с-1, поглощение осуществляется за 10-15—10-16 с (время, эквивалентное прохождению одной световой волны). За этот период ядра успевают сместиться лишь весьма незначительно, поскольку частота их колебаний много меньше указанной величины. Принцип Франка — Кондона гласит, что за время электронного перехода никаких существенных изменений в положениях атомных ядер молекулы не происходит. Рассмотрим рис. 13-5, на котором изображены два типа кривых потенциальной энергии молекул в возбужденном состоянии [5]. В первом случае геометрия молекулы в основном и возбужденном состояниях почти одинакова. Важно помнить, что при комнатной температуре большая часть молекул находится на самых нижних энергетических уровнях, по крайней мере для большинства колебательных состояний ![]()
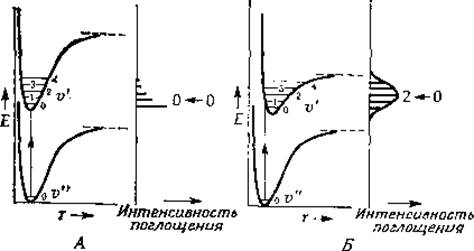
РИС. 13-5. Типичные кривые потенциальной энергии для полосатых спектров двух типов. А. Равновесные межъядерные расстояния rе в основном и возбужденном состояниях примерно одинаковы. Б. r'е (возбужденное состояние) >rе (основное состояние). ([5], стр. 179).
Таким образом, в случае рис. 13-5, A наиболее вероятными будут переходы с самых нижних колебательных подуровней основного электронного состояния. Наиболее вероятное расстояние между ядрами двухатомной молекулы в основном состоянии равно равновесному значению rе (рис. 13-2). Поскольку это расстояние для всех колебательных подуровней возбужденного электронного состояния одинаково, переход может произойти в любое из этих состояний, но наиболее вероятен переход на первый колебательный подуровень возбужденного состояния. В результате мы имеем спектр поглощения, в котором наряду с интенсивной резкой линией, соответствующей «0—0-переходу», имеются более слабые линии, отвечающие переходам 0—1, 0—2, 0—3 и т. д. (рис. 13-5, A).
Спектр второго типа представлен на рис. 13-5, Б. В этом случае расстояние между ядрами за время перехода молекулы в возбужденное состояние увеличивается — rе оказывается больше, чем в основном состоянии. Согласно принципу Франка — Кондона, наиболее вероятным будет переход на те колебательные подуровни возбужденного состояния, для которых межъядерное расстояние большую часть времени примерно равно значению rе в основном состоянии. Из рис. 13-5,5 ясно, почему 0—0-переходу в спектре поглощения в этом случае отвечает менее интенсивная линия, чем переходам на более высокие уровни.
Реально наблюдаемые спектры поглощения, в особенности спектры поглощения многоатомных молекул, имеют гораздо более сложную природу. Одним из факторов, усложняющих ситуацию, является то, что некоторые молекулы в основном состоянии находятся на более высоких колебательных подуровнях, соответствующих низкоэнергетическим типам колебаний. Поэтому в спектре появляются более слабые линии, расположенные также и с низкоэнергетической стороны от 0—0-перехода. Так как у многоатомных молекул существует несколько типов нормальных колебаний, между теми полосами, которые изображены на рис. 13-5, появляются другие. Все эти полосы уширяются из-за вращения молекул и взаимодействия их с растворителем.
Примером соединения, имеющего спектр такого типа, является толуол (метилбензол). Спектр паров толуола содержит большое число резких линий — некоторые из них видны
даже на спектре, полученном при низком разрешении (рис. 13-6). Несколько серий из последовательных линий удается идентифицировать [22]. Одна из них начинается с интенсивной линии, характеризующейся волновым числом 37,48 кК, которая отвечает 0—0-переходу; расстояние между соседними линиями, равное ~930 см-1, соответствует колебаниям, приводящим к симметричному расширению и сжатию («дыханию») кольца; эти колебания можно обнаружить, сняв инфракрасный спектр соединения. Другие последовательности, начинающиеся с линии, которая отвечает 0—0-переходу, соответствуют колебаниям с частотами (в возбужденном электронном состоянии) 460, 520 и 1190 см-1. Более слабые полосы на спектре, представленном на рис. 13-6, «скрыты» в областях между максимумами.
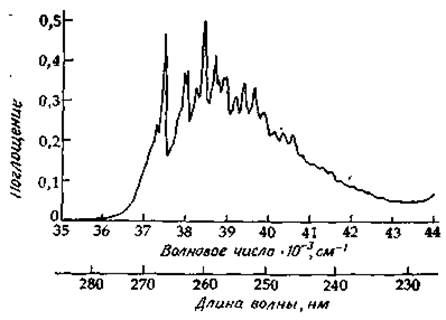
РИС. 13-6. Спектр паров толуола, соответствующий переходу в первое электронно-возбужденное состояние; спектр снят при низком разрешении на спектрофотометре Кэри 1501.
При снятии спектра поглощения толуола в растворе резкие линии уширяются, но колебательная структура полностью не исчезает. Как видно из рис. 13-7, спектры фенилаланина и его производных [22] очень похожи на спектр толуола (0—0-переходу соответствует частота 37 310 см-1); колебательная структура спектра фенилаланина четко проявляется в спектрах многих белков (см., например, рис. 13-14). Аналогичный вид имеет и спектр поглощения тирозина (рис. 13-7), но при этом линия, отвечающая 0—0-переходу, сдвинута в сторону более низких энергий (~ 35 300 см-1 в воде). Четко видны последовательности линий с интервалами 1200 и 800 см-1 [23].
Для описания формы одной электронной полосы в качестве огибающей ее колебательных компонентов часто используют гауссову кривую (нормальное распределение). В некоторых случаях, например для медьсодержащего голубого белка из Pseudomonas (рис. 13-8), представление полос в виде гауссовых кривых оказывается вполне адекватным и позволяет разложить спектр на компоненты, отвечающие конкретным электронным переходам. Каждый переход характеризуют положением максимума, его высотой (молярной экстинкцией) и шириной (измеряемой на уровне полувысоты в см-1). Однако полосы поглощения органических соединений, как правило, несимметричны — они растянуты в сторону более высоких энергий1). Для описания этих полос больше подходит несимметричная функция, например логарифмически-нормальное распределение [24, 25]. Помимо положения пика, его высоты и ширины вводится четвертый параметр, являющийся мерой асимметричности пика. Подбор логарифмически-нормальных кривых с помощью ЭВМ позволяет точно указать положение пиков, их ширину и амплитуду. Заметим, что такой способ локализации пиков обычно приводит к небольшому смещению линии, отвечающей 0—0-переходу, в сторону более высоких энергий. Ширина пиков может быть различной, но чаще всего составляет 3000—4000 см-1.
1) Спектры поглощения представляют в виде функций от длины волны. Хотя гауссовы кривые иногда довольно хорошо вписываются в эти спектры, ширину полос в нанометрах выражать нежелательно. Величиной, пропорциональной энергии, является волновое число. Если изображать спектральные полосы в виде зависимости поглощения не от длины волны, а от волнового числа, то в видимой и ультрафиолетовой областях их ширина будет примерно одинаковой.
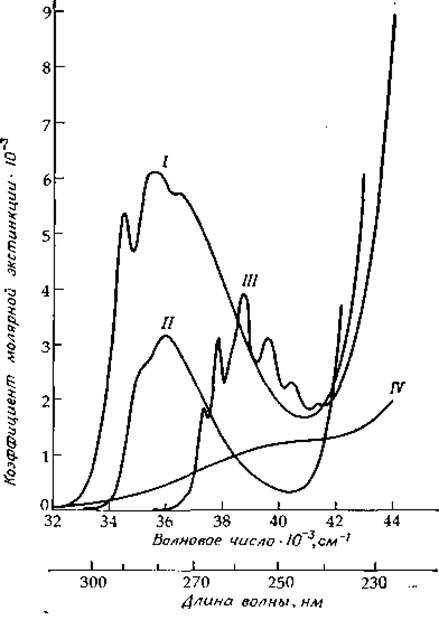
РИС. 13-7. Спектры поглощения N-ацильных производных этиловых эфиров триптофана (I), тирозина (II), фенилаланина (III) и диметилового эфира цистина (IV) в метаноле при 25 °С, соответствующие переходам указанных соединений в первое электронно-возбужденное состояние. Спектры производных тирозина, фенилаланина и цистина умножены на коэффициенты 2, 20 и 4 соответственно [41].
Другой ценный подход к количественному анализу спектров основан на описании каждой колебательной полосы своей гауссовой кривой [26,27].
б. Классификация переходов
Интенсивную полосу поглощения в области 600 нм в спектре медьсодержащего белка, раствор которого имеет голубую окраску (рис. 13-8), приписывают- d—d-переходу электрона, принадлежащего иону металла [28]. Высокая интенсивность полосы может быть обусловлена наличием связи между ионом металла и атомом S остатка метионина, входящего в состав белка [29]. Электронные переходы у большинства органических молекул относятся к другому типу. Переходы с частотами <55 000 см-1 классифицируются либо как n—п*; либо, как n—п*-переходы. В первом из указанных случаев электрон переходит со связывающей п-орбитали на разрыхляющую п*-орбиталь. В этилене такой переход наблюдается при частоте 61 540 см-1 (162,5 нм); максимальное значение молярной экстинкции εmax составляет ∼15 000 М-1∙см-1. n—п*-Переходы обусловлены перемещением электрона неподеленной пары атома кислорода или азота на разрыхляющую п*-орбиталь и выражены очень слабо. Например, n—п*-переход для ацетона в Н2О ![]() = 37 740 см-1, λmах = 265 нм) характеризуется значением εmах, равным ~240; ширина полосы составляет ~6400 см-1. Особенностью n—п*-перехода является сильное смещение полосы поглощения в сторону низких энергий при переносе соединения из воды в менее полярный растворитель. Так, максимум полосы поглощения ацетона в метаноле соответствует
= 37 740 см-1, λmах = 265 нм) характеризуется значением εmах, равным ~240; ширина полосы составляет ~6400 см-1. Особенностью n—п*-перехода является сильное смещение полосы поглощения в сторону низких энергий при переносе соединения из воды в менее полярный растворитель. Так, максимум полосы поглощения ацетона в метаноле соответствует ![]() см-1, а в гексане — 35 970 см-1 (278 нм). Подобный сдвиг под влиянием растворителя считают «опознавательным признаком» n—п*-перехода, часто полагая, что полосы, соответствующие : n—п*-переходам, при изменении характера, растворителя сдвигаются в противоположную сторону. Однако для многих полярных хромофоров, присутствующих в биохимических соединениях, это неверно. Так, n—п*-полосы тирозина при перенесении этого соединения из воды в гексан тоже смещаются к более низким энергиям. Правда, величина сдвига намного меньше, чем для n—п*-полосы ацетона.
см-1, а в гексане — 35 970 см-1 (278 нм). Подобный сдвиг под влиянием растворителя считают «опознавательным признаком» n—п*-перехода, часто полагая, что полосы, соответствующие : n—п*-переходам, при изменении характера, растворителя сдвигаются в противоположную сторону. Однако для многих полярных хромофоров, присутствующих в биохимических соединениях, это неверно. Так, n—п*-полосы тирозина при перенесении этого соединения из воды в гексан тоже смещаются к более низким энергиям. Правда, величина сдвига намного меньше, чем для n—п*-полосы ацетона.

РИС. 13-8. Разложение спектров КД (А) и поглощения (Б) голубого белка из Pseudomonas, лежащих в видимой области, на несколько перекрывающихся гауссовых кривых, которые соответствуют отдельным спектральным полосам (штриховые линии). Номерами от 1 до 6 обозначены полосы, занимающие в обоих спектрах одинаковые положения и имеющие одинаковую ширину. Сплошные линии — результат сложения гауссовых кривых. Каждая такая огибающая в пределах ошибки измерений совпадает с экспериментально снятыми спектрами. Штрих-пунктирная часть огибающей на спектре КД выше 700 нм вычерчена по форме полосы I в спектре поглощения [28].
Молекула может переходить не только на первый, но и на более высокие энергетические уровни. Так, у бензола и его производных легко обнаруживаются три п—п*-перехода (рис. 13-9). Первый представлен слабой полосой с ε = 102—103. Вторая полоса отвечает более высокой частоте (в 1,35±0,10 раз выше частоты первой полосы) и характеризуется εmах до 104. Третья полоса соответствует еще более высокой энергии, а εmах достигает значения 5∙104. Энергетические уровни, отвечающие этим переходам, согласно часто применяемой системе обозначений Платта, записываются как 1Lb, 1La и 1Ва. Другие авторы описывают эти уровни, опираясь на симметрию молекулярных орбиталей. Так, основное состояние обозначается 1Alg, а три возбужденных — как 1В2u 1В1u и 1E1u. Индекс 1 указывает, что рассматриваемое возбужденное состояние является синглетным, т. е. что электроны в возбужденном состоянии остаются спаренными (поглощение видимого и ультрафиолетового света почти всегда переводит молекулу в синглетное возбужденное состояние). Для более сложных циклических систем число возможных переходов возрастает. Зачастую эти переходы пытаются сопоставить с переходами в бензоле.
Интенсивности, соответствующие электронным переходам, сильно различаются между собой. Площадь полосы поглощения (A) на графике зависимости ε от волнового числа ![]() прямо пропорциональна безразмерной величине, называемой силой осциллятора f:
прямо пропорциональна безразмерной величине, называемой силой осциллятора f:
![]()
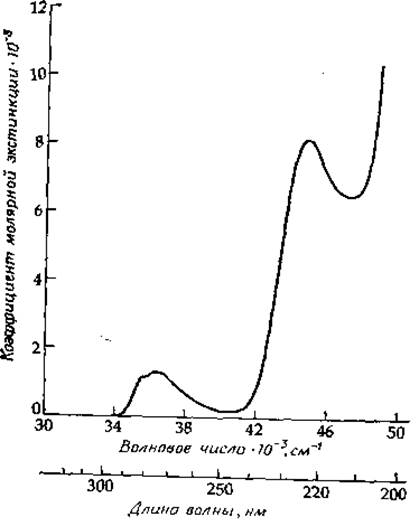
РИС. 13-9. Спектр этилового эфира N-ацетилтирозина в водном фосфатном буфере, pH 6,8. Обратите внимание на три n—п*- перехода возрастающей интенсивности. Третьему n—п*-переходу ароматического кольца соответствует vmax≃52 000 см-1; при этом коэффициент молярной экстинкции достигает значения ~ 40 000. В поглощение в высокоэнергетической части спектра дают вклад также n—п*- и n—п*-переходы амидной группы этого соединения.
В этом уравнении mе и е — масса и заряд электрона соответственно, с — скорость света, N — число Авогадро, A — площадь полосы на графике зависимости є от ![]() в см-1; F представляет собой безразмерный поправочный множитель, связанный с показателем преломления среды; для водных растворов он очень близок к единице. Если полосу поглощения представить в виде треугольника с высотой εmах и основанием, равным ширине полосы W (измеряемой на уровне полувысоты), то для типичной полосы поглощения С εmах = 104 и W = 3000 см-1 получим f = 0,13.
в см-1; F представляет собой безразмерный поправочный множитель, связанный с показателем преломления среды; для водных растворов он очень близок к единице. Если полосу поглощения представить в виде треугольника с высотой εmах и основанием, равным ширине полосы W (измеряемой на уровне полувысоты), то для типичной полосы поглощения С εmах = 104 и W = 3000 см-1 получим f = 0,13.
Согласно теории поглощения, сила осциллятора связана с вероятностью перехода и приближается к единице лишь для самых сильных электронных переходов. Такой высокой сила осциллятора бывает очень редко. Например, для Сu2+ она равна ~10-4, а для полосы поглощения толуола, представленной на рис. 13-6, ∼ 2∙10—3. Низкая интенсивность полос поглощения производных бензола определяется тем обстоятельством, что для идеально симметричных молекул эти переходы являются запрещенными. Переход 1Lb для бензола становится слабо разрешенным лишь вследствие сопряжения с асимметричными колебаниями кольца. В спектре бензола линия, соответствующая переходу 0—0, отсутствует; разрешены лишь последующие линии, отвечающие дополнительному поглощению энергии несимметричных колебаний, равной 520 см-1. Благодаря асимметрии колец толуола и фенилаланина, обусловленной наличием в них замещающих групп, 0—0-переход становится разрешенным и сила осциллятора принимает более высокое значение, чем у бензола. 1Lа-переход бензольных производных также частично запрещен правилами отбора, и лишь для третьей полосы сила осциллятора приближается к единице.
в. Поляризация переходов
Вероятность перехода прямо связана с дипольным моментом перехода (или просто с моментом перехода) — векторной величиной, зависящей от дипольного момента молекулы в основном и возбужденном состояниях. Для ароматических циклических систем векторы дипольных моментов n—п*-переходов лежат в плоскости кольца. Однако их направление и величина для различных n—п*-переходов оказываются разными.
Дипольный момент перехода имеет размерность длины (обычно его выражают в ангстремах); его можно представить как меру смещения зарядов в процессе перехода. Свет наиболее эффективно поглощается в том случае, когда направление его поляризации (т. е. направление вектора напряженности электрического поля) и направление момента перехода совпадают. В этом легко убедиться, измеряя поглощение света кристаллами. Как и инфракрасные спектры поглощения ориентированных пептидных цепей (рис. 13-3), электронные спектры кристаллов обнаруживают четко выраженный дихроизм.
В отличие от n—п*-переходов n—п*-переходы в гетероциклических соединениях и карбонилсодержащих кольцах часто поляризованы в направлении, перпендикулярном плоскости кольца.
г. Связь максимума полосы поглощения и ее интенсивности со структурой соединения
Хотя квантовомеханические расчеты позволяют предсказать число полос поглощения и приблизительно указывают их местоположение, они не дают необходимой точности при расшифровке спектров. Поэтому в электронной спектроскопии широко используются эмпирические правила и атласы спектров, позволяющие проводить сравнительный анализ [30, 31]. Ориентироваться в этой области читателю помогут следующие указания. Положение полосы поглощения сдвигается батохромно (в сторону более длинных волн, более низких энергий) при увеличении числа сопряженных двойных связей. Так, максимум поглощения бутадиена соответствует 46100 см-1 (217 нм), а этилена — 61500 см-1. По мере дальнейшего возрастания числа двойных связей батохромний сдвиг становится все меньше и меньше (но остается почти постоянным, если измерять его не в волновых числах, а в длинах волн). Для ликопена (рис. 12-14), содержащего 11 сопряженных двойных связей, полоса поглощения находится при 21 300 см-1 и имеет отчетливо выраженную колебательную структуру (рис. 13-10). Спектры некоторых циклических молекул, например порфиринов и хлорофиллов, можно соотнести со спектрами линейных полиенов. Отметим (рис. 10-2), что а- и ß-полосы порфирина являются компонентами колебательной структуры одного и того же электронного перехода, тогда как интенсивная полоса Соре порождается другим переходом.
Полосы поглощения замещенных бензольных колец почти всегда сдвинуты в сторону более низких энергий относительно полосы поглощения исходного углеводорода. Чем сильнее выражена способность замещающих групп оттягивать на себя или отдавать электроны, тем больше батохромный сдвиг. Величина сдвига коррелирует с постоянной Гаммета σ. Так, первая полоса поглощения тирозина в воде смещена на 2600 см-1 в красную сторону от полосы бензола, тогда как для диссоциированного тирозинового аниона сдвиг составляет 4700 см-1 — в очень грубом приближении сдвиг действительно пропорционален σр (табл. 3-9). Особенно большой сдвиг наблюдается в тех случаях, когда в одном и том же кольце присутствуют противоположные по характеру функциональные группы (например, электронодонорные и электроноакцепторные). Эффект пар заместителей в орто- и мета-положениях примерно одинаков (в отличие от влияния этих заместителей на реакционную способность). Когда замещающие группы находятся в пара-положении, спектральные сдвиги оказываются несколько иными. При наличии более чем двух замещающих групп характер спектра определяется главным образом двумя группами, оказывающими наиболее сильное влияние. Полезные эмпирические правила можно найти в работах [32] и [33].
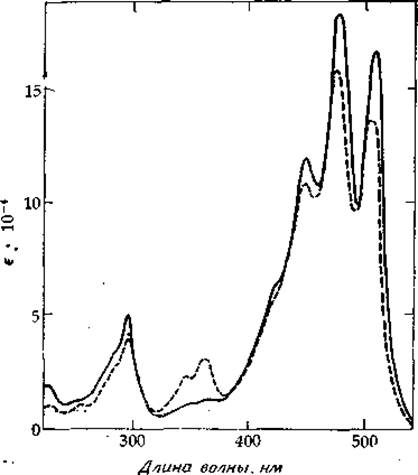
РИС. 13-10. Спектр поглощения ликопена. Обратите внимание на его колебательную структуру в области ∼1200—1500 см-1.
Сплошная линия соответствует полностью транс-ликопену, a штриховая — тому же образцу после выдерживания в течение 45 мин в темноте. Обратите внимание на появление максимума при ~ 360 нм, обусловленного образованием изомеров, в которых имеются двойные цис-связи.
д. Спектры нуклеиновых кислот и белков
Большинство белков имеет интенсивную полосу поглощения с максимумом 280 нм (35 700 см-1), что обусловлено присутствием ароматических аминокислот триптофана, тирозина и фенилаланина [34]. Форма полосы с достаточной степенью точности определяется формой полос поглощения указанных ароматических аминокислот с учетом содержания последних в белке. Спектры поглощения простых амидных производных фенилаланина, тирозина и триптофана в этой области представлены на рис. 13-7 и 13-9. Низкоэнергетическая полоса триптофана соответствует двум перекрывающимся переходам 1La и 1Lb [26]. Полоса, соответствующая 1Lb-переходу, имеет четко выраженную колебательную структуру, тогда как полоса 1La носит более диффузный характер. Максимум 0—0-полос для обоих указанных переходов у производных триптофана при растворении их в углеводородных растворителях равен ∼289,5 нм (34 540 см-1). Однако в белках 1La-полоса может быть смещена на 3—10 нм (на ∼1100 см-1) в сторону более низких энергий. Этот сдвиг является, по-видимому, результатом образования водородных связей с другими группами белка. Самый большой сдвиг наблюдается в тех случаях, когда NH-группа индольного кольца образует водородную связь с группой СОО-, с кольцевым атомом азота гистидина или с карбонильной группой амидов [35]. В водной среде 1Lb-полоса поглощения триптофана смещается в сторону более высоких, а 1Lа-полоса — в сторону более низких энергий относительно соответствующих полос в случае углеводородного растворителя. Из рис. 13-7 можно заметить, что вклад остатков триптофана в поглощение света белками намного превосходит вклад эквивалентного числа остатков тирозина или фенилаланина. Таким образом, для большинства белков поглощение триптофана является определяющим.
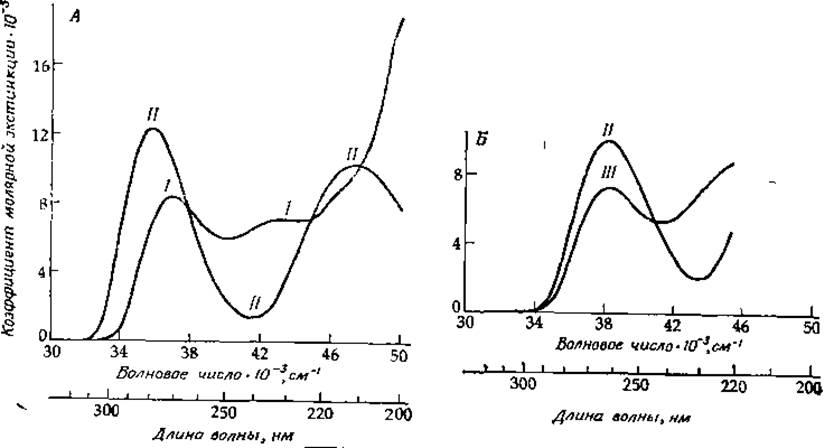
РИС. 13-11. Спектры поглощения цитидина (А) и уридина (Б) в ближней ультрафиолетовой области. I — монопротонированная форма цитидина (рКа = 4,2); II — нейтральные формы (рН~7); III — моноанионная форма уридина (рКа = 9,2).
Помимо трех указанных ароматических аминокислот в ближней ультрафиолетовой области поглощают дисульфидные связи (рис. 13-7). Поскольку характеристики этого процесса зависят от величины двугранных углов, образуемых дисульфидными мостиками, определить вклад данного хромофора в полосу с λmах = 280 нм довольно трудно.
Следует иметь в виду, что тирозин, триптофан и фенилаланин имеют также полосы поглощения в высокоэнергетической части УФ-спектра белков. Еще больший вклад в поглощение белков в этой области вносят амидные группы; вклад становится ощутимым при значениях больше 45 000 см-1. Здесь наблюдается слабый n—п*-переход с Vmax = 47 500 см-1 (λmах = 210 нм), перекрывающийся с сильным n—п*- переходом c λmах ≃ 52 600 cм-1 (λmах = 190 нм). В этой области находятся также полосы поглощения гистидина.
Как и в случае полипептидов, свойства спектров поглощения полинуклеотидов отражают спектральные свойства их компонентов. На рис. 13-11 и 13-12 приведены спектры поглощения пуриновых и пиримидиновых рибонуклеозидов. Число отдельных электронных переходов точно не известно, а их природа далеко не очевидна, несмотря на многочисленные попытки расшифровать эти спектры и соотнести их между собой [36]. То же самое можно сказать и о флавинах, спектры поглощения которых содержат по меньшей мере четыре интенсивные полосы перехода (рис. 8-16) [37].
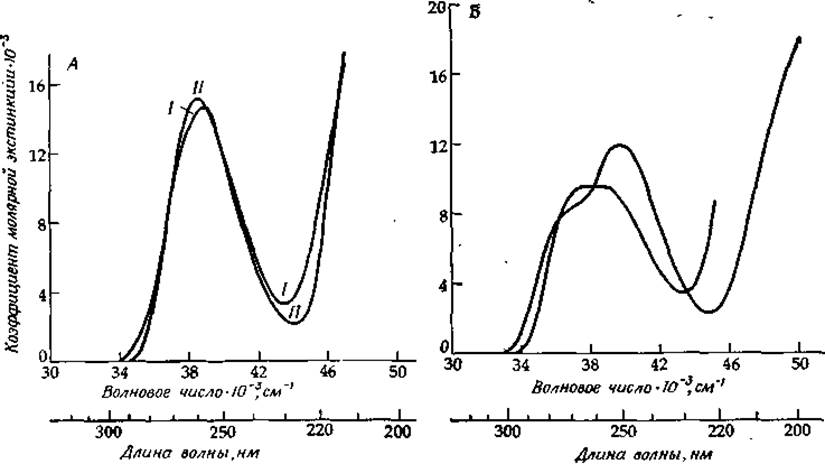
РИС. 13-12. УФ-спектры поглощения аденозина (Л) и гуанозина (Б). I — монопротонированная форма аденозина (рКа = 3,5); II— нейтральные формы; III — моноанион гуанозина (рКа = 9,2).
Если в белках максимум низкоэнергетической полосы поглощения соответствует λ ≃ 280 нм, то для полинуклеотидов λmax = 260 нм (38 500 см-1). При исследовании оптических свойств нуклеиновых кислот особенно важной характеристикой является гипохромный эффект. В то время как поглощение денатурированного полинуклеотида примерно равно суммарному поглощению его компонентов, при образовании двухцепочечной структуры с укладкой оснований одно над другим поглощение при 260 нм уменьшается на 34%. Это явление лежит в основе оптического метода исследования плавления полинуклеотидов (рис. 2-28). Физическая природа гипохромного эффекта кроется во взаимодействии тесно уложенных одно над другим пар оснований (стэкинг-взаимодействие) [38].
е. Разностная спектроскопия
Часто бывает необходимо исследовать изменения поглощения света белками или нуклеиновыми кислотами в зависимости от таких факторов, как pH, температура, ионное окружение и присутствие или отсутствие других взаимодействующих молекул. Поскольку происходящие при этом изменения спектров невелики, широко практикуется измерение разности двух спектров — «невозмущенного» спектра и спектра, снятого в присутствии «возмущающего агента». В роли последнего может выступать какой-либо реагент, добавленный в раствор (например, глицерин, D2O и т. д.), а также pH и температура. Разностный спектр, представленный на рис. 13-13,5, порожден связыванием каталитической субъединицы аспартат — карбамоилтрансферазы (гл. 4, разд. Г, 8) с ингибитором сукцинатом и субстратом карбамоилфосфатом [39]. Этот спектр характеризуется присутствием в области поглощения ароматических аминокислот двух пиков и широкого минимума. При надлежащей интерпретации (непременное условие!) такие разностные спектры могут дать представление об изменении в окружении ароматических аминокислот, входящих в состав данного белка [40].
Разностные спектры обычно регистрируют, пропуская два световых пучка через две строго выверенные кюветы: один из пучков — через кювету сравнения, другой — через кювету с образцом. Однако спектр, приведенный на рис. 13-13,5, получен путем независимой регистрации двух спектров (с выводом данных на перфокарты) и последующего вычитания их один из другого с помощью вычислительной машины. Те же данные могут быть представлены и иначе: в каждую из полос поглощения вписывают логарифмически-нормальную кривую (разд. Б, 4, а), а затем наносят на график разность между сглаженной кривой и экспериментальными точками, следующими с очень малым интервалом [41], как это показано на рис. 13-13,5. Два графика, полученные таким образом, выявляют тонкую структуру спектра. Это по существу иной способ получения разностного спектра. Преимуществом указанного метода служит то, что проведенная с помощью ЭВМ аппроксимация кривой дает представление о форме самой полосы поглощения. Как нетрудно видеть, связывание сукцината и карбамоилфосфата вызывает слабый сдвиг (20 см-1) полосы поглощения и очень небольшое ее уширение. Основной эффект состоит в более четком выражении колебательной структуры 0—0-полосы при 34 600 см-1, обусловленной поглощением двух остатков триптофана, которые присутствуют в субъединице фермента. Причина этого изменения, однако, не вполне понятна, в чем и проявляется ограниченность метода разностной спектроскопии.