БИОХИМИЯ - Л. Страйер - 1984
ТОМ 2
ЧАСТЬ II ГЕНЕРИРОВАНИЕ И ХРАНЕНИЕ МЕТАБОЛИЧЕСКОЙ ЭНЕРГИИ
ГЛАВА 16. ГЛИКОГЕН И ОБМЕН ДИСАХАРИДОВ
16.18. Обмен гликогена в печени регулирует содержание глюкозы в крови
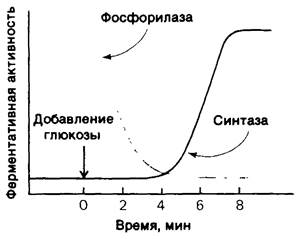
Контроль синтеза и распада гликогена в печени занимает центральное место в регуляции содержания глюкозы в крови. В норме этот уровень колеблется оттот 80 до 120 мг на 100 мл. Печень чувствительна к концентрации глюкозы в крови: если содержание глюкозы в крови превышает пороговый уровень, печень поглощает глюкозу; если же ее содержание ниже этого уровня, печень высвобождает глюкозу. Количество фосфорилазы а в печени быстро уменьшается при вливании глюкозы (рис. 16.10). После лаг-периода возрастает количество гликоген-синтазы а, что приводит к синтезу гликогена. Недавно было установлено, что в клетках печени фосфорилаза служит глюкозным датчиком-чувствительным элементом для глюкозы. Связывание глюкозы с фосфорилазой а сдвигает аллостерическое равновесие из R-состояния в Т-состояние (см. рис. 16.5). В результате фосфорильная группа при серине-14 становится доступной для гидролиза фосфатазой. Значительную роль играет при этом то обстоятельство, что фосфатаза, тесно связываясь с фосфорилазой а, проявляет свое каталитическое действие только после перехода последней в Т-состояние под действием глюкозы.
Каким образом осуществляется активация гликоген-синтазы глюкозой? Напомним, что одна и та же фосфатаза действует на фосфорилазу и на гликоген-синтазу. Фосфорилаза b в отличие от а-формы не связывает фосфатазу. Следовательно, превращение фосфорилазы а в фосфорилазу b сопровождается освобождением фосфатазы, которая теперь может использоваться для активации гликоген-синтазы. Удаление фосфорильной группы из неактивной синтазы b превращает ее в активную а-форму. Вначале на одну молекулу фосфатазы приходится около десяти молекул фосфорилазы а. Следовательно, повышение активности синтазы может начаться только после перехода большей части фосфорилазы а в форму b(рис. 16.10). Эта замечательная, чувствительная к глюкозе система зависит от ключевых элементов: 1) связи между аллостерическим центром для глюкозы и серинфосфатом, 2) использования одной и той же фосфатазы для инактивации фосфорилазы и активации гликогенсинтазы и 3) связывания фосфатазы с фосфорилазой а для предотвращения преждевременной активации гликоген-синтазы.
Рис. 16.10. Инфузия глюкозы приводит к инактивации фосфорилазы, которая сопровождается активацией гликоген-синтазы
16.19. Известен ряд генетически детерминированных болезней накопления гликогена
Первая болезнь накопления гликогена была описана Эдгаром фон Гирке (Edgar von Gierke) в 1929 г. У больного был огромный живот вследствие массивного увеличения печени. Отмечалась выраженная гипогликемия между приемами пищи. Кроме того, после введения адреналина и глюкагона содержание сахара в крови не повышалось. У детей с этим заболеванием могут наблюдаться судороги, связанные с низким содержанием глюкозы в крови.
Природу ферментативного нарушения при болезни Гирке раскрыли Кори в 1952 г. Они установили, что в печени таких больных отсутствует глюкозо-6-фосфатаза. Это был впервые обнаруженный случай врожденной недостаточности фермента печени. Гликоген печени у таких больных имел нормальную структуру, но присутствовал в необычно больших количествах. Отсутствие глюкозо-6-фосфатазы в печени вызывает гипогликемию, потому что не происходит образования глюкозы из глюкозо-6-фосфата. Фосфорилированный же сахар не выходит из печени, поскольку он не может пересечь плазматическую мембрану. Происходит компенсаторное усиление гликолиза в печени, обусловливающее повышенное содержание лактата и пирувата в крови. Для людей с болезнью Гирке характерна также повышенная зависимость от жирового обмена.
К настоящему времени уже охарактеризован целый ряд болезней накопления гликогена (табл. 16.1). Кори раскрыли природу биохимического дефекта и при другой болезни накопления гликогена (тип III), которую нельзя отдифференцировать от болезни Гирке (тип I) путем одного только медицинского обследования. Болезнь типа III характеризуется аномальной структурой гликогена мышц и печени и значительным увеличением его количества. Самым резким отклонением от нормы является очень маленькая длина внешних ветвей гликогена. У таких больных отсутствует фермент, разрывающий связи в местах ветвления, (α-1,6-глюкозидаза), и поэтому у них могут эффективно использоваться только самые удаленные от центра молекулы ветви гликогена. Следовательно, лишь небольшая часть этого аномального гликогена функционально активна в качестве доступной резервной формы глюкозы.
Таблица 16.1. Болезни накопления гликогена
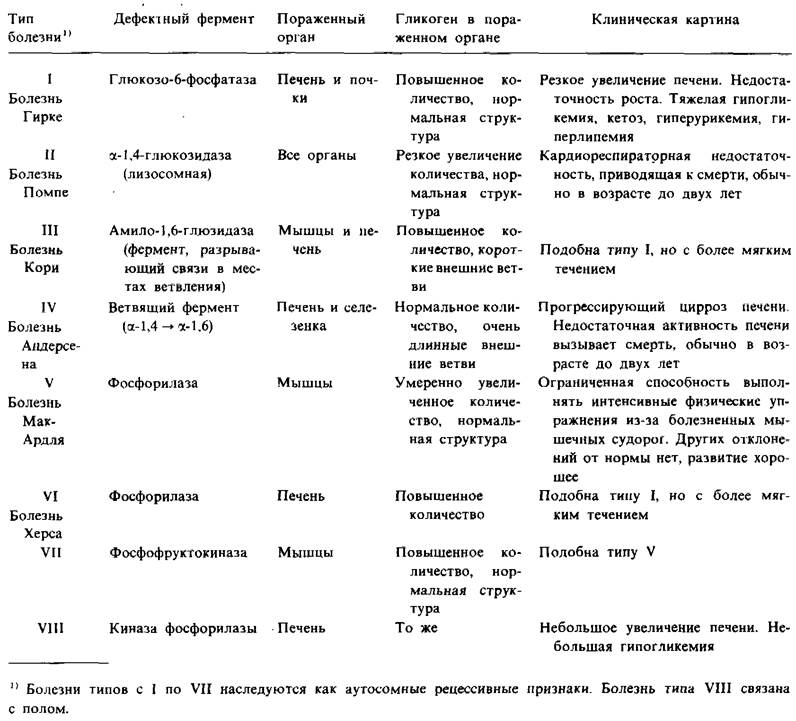
Нарушение обмена только мышечного гликогена имеет место при болезни Мак- Ардля (МсАгdlе) (тип V). В этом случае в мышцах отсутствует фосфорилазная активность, и больные обладают ограниченной способностью к выполнению интенсивных физических упражнений из-за болезненных мышечных судорог. Других отклонений от нормы не обнаруживается, больные хорошо развиваются. Таким образом, эффективное использование мышечного гликогена не является жизненно необходимым.
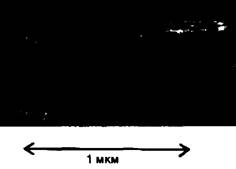
Рис. 16.11. Электронная микрофотография скелетной мышцы ребенка с болезнью накопления гликогена типа II (болезнь Помпе). Лизосомы перегружены гликогеном вследствие недостаточности α-1,4-глюкозидазы. Этот дефект расщепления гликогена ограничен лизосомами. Количество гликогена в цитозоле находится в норме
16.20. Крахмал-резервный полисахарид у растений
Мы обратимся теперь к другим распространенным полисахаридам. Резервным полисахаридом у растений является крахмал, который существует в двух формах. Амилоза, неразветвленный тип крахмала, состоит из глюкозных остатков, соединенных α-1,4-связью. В амилопектине, разветвленной форме, на тридцать α-1,4-связей имеется примерно одна α-1,6-связь. Таким образом, он подобен гликогену, отличаясь от последнего более низкой степенью ветвления.
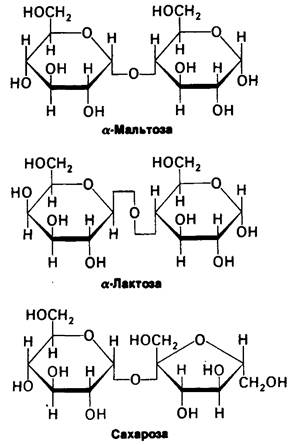
На долю крахмала приходится больше половины потребляемых человеком углеводов. И амилопектин, и амилоза быстро гидролизуются α-амилазой, которая секретируется слюнными железами и поджелудочной железой. Альфа-амилаза гидролизует внутренние α-1,4-связи, приводя к образованию мальтозы, мальтотриозы и α-декстрина. Мальтоза состоит из двух остатков глюкозы, соединенных между собой α-1,4-связью (рис. 16.12); мальтотриоза состоит из трех таких остатков. Альфа-декстрин образован несколькими остатками глюкозы, соединенными между собой α-1,6-связью в дополнение к α-1,4-связям. Мальтоза и малтотриоза гидролизуются до глюкозы под действием мальтазы, тогда как α-декстрин гидролизуется до глюкозы α-декстриназой. В солоде присутствует другой вид амилазы, называемый β-амилазой; она гидролизует крахмал до мальтозы. Бета-амилаза действует только на остатки невосстанавливающего конца.
Рис. 16.12. Строение наиболее широко распространенных дисахаридов: α-мальтозы, α-лактозы и сахарозы. Буква α относится к конфигурации аномерного углеродного атома на восстанавливающем конце дисахарида, а не к конфигурации гликозидной связи
Другой главный полисахарид растений, это целлюлоза, выполняющая не питательную, а структурную функцию. В действительности целлюлоза является наиболее распространенным органическим соединением в биосфере, содержащим более половины всего органического углерода. Она представляет собою неразветвленный полимер из остатков глюкозы, соединенных β-1,4-связями. У млекопитающих нет целлюлаз, и поэтому они не способны переваривать древесину и растительные волокна. Однако в пищеварительном тракте некоторых жвачных обитают бактерии, продуцирующие целлюлазу, благодаря чему эти животные могут переваривать целлюлозу.
Декстрин-другой полисахарид, состоящий только из остатков глюкозы, соединенных между собой преимущественно α-1,6-связью. Отдельные ветви в зависимости от вида организма образуются α-1,2-, α-1,3- или α-1,4-связями. Декстран служит резервным полисахаридом у дрожжей и бактерий. Наружный скелет (экзоскелет) насекомых и ракообразных содержит хитин, построенный из остатков N-ацетил- глюкозамина при помощи β-1,4-связей. Хитин, таким образом, подобен целлюлозе, с той лишь разницей, что в нем заместителем при С-2 служит ацетилированная аминогруппа, а не гидроксильная группа.
16.21. Мальтоза» сахароза и лактоза- широко распространенные дисахариды
Структура этих трех распространенных дисахаридов показана на рис. 16.12. Как упоминалось выше, мальтоза возникает в результате гидролиза крахмала и затем гидролизуется мальтазой до глюкозы. Сахарозу, обычный столовый сахар, получают промышленным способом из сахарного тростника или сахарной свеклы, Аномерные углеродные атомы остатков глюкозы и фруктозы связаны в сахарозе α-гликозидной связью. Поэтому в сахарозе в отличие от других сахаров отсутствует восстанавливающая концевая группа. Гидролиз сахарозы до глюкозы и фруктозы катализируется сахарозой. Лактоза представляет собой дисахарид, содержащийся
в молоке, и нигде больше в сколько-нибудь заметном количестве не обнаружена. Она гидролизуется лактазой с образованием галактозы и глюкозы. Лактаза, сахараза, мальтаза и α-декстриназа связаны с клетками слизистой оболочки тонкого кишечника.
UPD-сахара - активированные промежуточные продукты при синтезе сахарозы и лактозы, так же как UPD-глюкоза служит донором глюкозильного остатка при синтезе гликогена. На самом деле нуклео- зиддифосфатсахара являются активированными промежуточными продуктами почти во всех процессах синтеза гликозидных связей. Например, целлюлоза в зависимости от вида растений синтезируется из аденозиндифосфоглюкозы (АDР-глюкозы), цитидиндифосфоглюкозы (СDР- глюкозы) или гуанозиндифосфоглюкозы (GDР-глюкозы). Сахароза синтезируется путем переноса глюкозы от UDР-глюкозы на фруктозо-6-фосфат с образованием сахарозо-6-фосфата, который затем гидролизуется до сахарозы.