Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Примеры: предсказание функции структур, полученных в проектах по структурной геномике
Несколько особых примеров
Хотя имеется относительно немного масштабных работ, есть большое число отдельных интересных структур, опубликованных различными консорциумами по структурной геномике или в сотрудничестве с ними, где наличие структуры белка оказывалось принципиально важным для описания его функции. Одним из таких примеров является белок Тт0936 из Thermotoga maritima (имеющий PDB код 2рlm), который был получен в сторонней лаборатории с использованием клонов, предоставленных Объединенным Центром по Структурной Геномике. Белок был аннотирован в базах данных, как белок с неизвестной функцией, принадлежащий семейству амидогидролаз по классификации Pfam (PF01979), которое содержит ряд деаминаз и, в свою очередь, является частью более широкого надсемейства амидогидролаз. Как пример белка с неизвестной функцией Тт0936 был отобран для анализа с помощью сервера ProFunc, и в результате обнаружилось возможное сходство с одним из 189 используемых сервером известных шаблонов ферментативных активных центров.
Схожим активным центром (с математическим ожиданием случайности сходства Е = 2.45 х 1(Г4) оказался шаблон активного центра аденози- новой деаминазы (КФ 3.5.4.4), полученный из структуры с PDB кодом 1а41, которая участвует в пуриновом метаболизме. Общая идентичность последовательностей между этой структурой и Тт0936, вычисленная по парному выравниванию с помощью программы FASTA, составила лишь 24%, хотя структурное сходство (рассчитанное для структурного выравнивания сервером ProFunc, как доля пар остатков, лежащих в одном или нескольких совмещаемых сегментах, в общем числе эквивалентных остатков в выравнивании) достигает 95%, что говорит само за себя. Внутри сферы с радиусом 10 Å вокруг шаблона активного центра локальная идентичность последовательностей составляет 27,7%, что выше среднего по структуре и свидетельствует о большем сходстве между последовательностями в области вокруг активного центра. Кроме сильного сходства с аденозиновой деаминазой, было обнаружено и несколько совпадений обратных шаблонов с другими деаминазами и амидогидролазами.
Авторы структуры Тт0936 опубликовали работу, в которой предсказывают для этой структуры функцию адениндеаминазы (Hermann et al. 2007). Использованный ими подход включал проведение молекулярного докинга высокоэнергетических метаболических интермедиатов в структуру белка на том основании, что докинг исходных субстратов или продуктов может быть не столь эффективным, как докинг интермедиатов, стабилизированных ферментом. Получившийся список потенциальных лигандов состоял преимущественно из аналогов аденина, которые оказались хорошо подходящими для С6-деаминирования. Четыре из этих лигандов были протестированы в качестве субстратов, причем три из них показали существенную каталитическую константу скорости. Была определена структура комплекса между Тm0936 и продуктом (S-инозилгомоцистеином), образовавшимся при деаминировании S-аденозилгомоцистеина, и обнаружено очень точное соответствие между этим лигандом, связанным с Тm0936, и дезоксикоформицином, аналогом инозина, связанным со структурой, послужившей шаблоном при определении структуры Tm0936 (PDB код 1а41) (Рис. 11.1)
Интересно, что анализ фолда с использованием программы MSDfold показывает сходство с различными амидогидролазами и гуанин/цитозин-деаминазами. Причина, по которой этот сервер не выделяет структуру аденозиндеаминазы, как наиболее похожую, кроется в том, что структура самого Тm0936 имеет некоторые украшения за пределами совпадающей области, и структурное сходство падает ниже критического уровня в 70% по числу совпадающих элементов вторичной структуры (Рис. 11.2). Это наблюдение подчеркивает эффективность локальных сравнений и служит хорошим примером, когда функция может быть точно определена исходя из совпадения с шаблоном фермента, которое осталось бы незамеченным при использовании одних лишь методов анализа последовательности. Другой интересный пример можно найти в недавней публикации Центра по структурной геномике на Среднем Западе (MCSG), где описываются структуры открытого (R) и закрытого (Т) состояний префенатдегидратазы (PDT) (Tan et al. 2008), проясняющие наше понимание аллостерической регуляции этого фермента с помощью L-фенилаланина и других аминокислот. Префенатдегидратаза (КФ 4.2.1.51) превращает префенат в фенилпируват при биосинтезе L-фенилаланина и играет ключевую роль в этом процессе у организмов, использующих шикиматный метаболический путь, что делает этот фермент незаменимым для микроорганизмов. У человека этот фермент не обнаружен, и это означает, что он может быть выбран в качестве возможной мишени при разработке противомикробных препаратов.
Структуры префенатдегидратазы, размещенные в PDB (коды 2qmw и 2qmx), происходят из двух различных организмов и являются первыми кристаллографическими структурами PDT в расслабленном (R) и возбужденном (Т) состояниях (из Staphylococcus aureus и Chlorobium tepidum, соответственно). Эти ферменты демонстрируют низкую идентичность последовательностей (27,3%), но одинаковую общую архитектуру и доменную организацию: оба фермента являются тетрамерами (образуя димеры димеров - см. изображения индивидуальных димеров на Рис. 11.3) и состоят из каталитического домена (домена PDT) и регуляторного домена (домена ACT). Опираясь на эти структуры префенат дегидратаз, авторы предположили, что активный центр этого фермента располагается в зазоре между двумя доменами PDT.
Это предсказание, сделанное на основе структурного сходства, подтверждается так же анализом последовательности и данными по мутагенезу. Множественное выравнивание последовательностей и картирование выявленных консервативных остатков показало, что эти остатки локализованы на дне расселины между субдоменами. Остатки, по данным мутагенеза критичные для активности префенатдегидратазы в Е. coli, оказались эквивалентными остаткам в расселине между двумя PDT субдоменами (Zhang et al. 2000). Дополнительные данные мутагенеза для префенатдегидратазы в Corynebacterium glutamicum подтвердили, что эквивалентные остатки участвуют в связывании субтрата и/или каталитической активности (Hsu et al. 2004). В итоге, данные подтверждают, что и расселина, и консервативные остатки в ней образуют активный центр префенатдегидратазы, при этом Т168 является наиболее вероятным ключевым каталитическим остатком.

Рис. 11.1. (Цветную версию рисунка см. на вклейке.) Соответствие ферментативного активного центра шаблону, иллюстрирующее перекрывание между связанным S-инозитол-гомоцистеином в Тm0936 (показан оранжевым; PDB код 2plm) и дезоксикоформицином, аналогом инозина, связанным с шаблоном, присутствующим в аденозиндеаминазе (показан фиолетовым, PDB код 1а41). Связанные атомы цинка показаны перекрывающимися сферами тех же цветов, что и соответствующие лиганды. Остатки Тm0936 и аденозиндеаминазы показаны синим и красным, соответственно
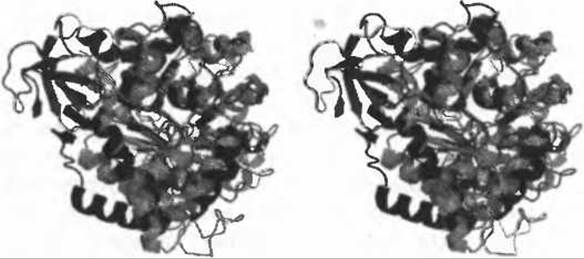
Рис. 11.2. Стереоизображение наложения рассматриваемой структуры Tm0936 (PDB код 2plm - показана черным) на структуру, использовавшуюся при создании шаблона ферментативного активного центра (PDB код 1а4l - показана серым). Дополнительные элементы вторичной структуры в рассматриваемом белке можно легко увидеть в левой части изображения
За определением вероятного активного центра последовало определение аллостерического сайта. Расположение этого сайта связывания L-фенилаланина в префенатдегидратазе сходно с расположением сайта связывания эффектора в некоторых других ферментах, имеющих домен ACT и участвующих в связывании аминокислот или других небольших молекул. Наличие связанного L-фенилаланина в структуре позволило визуализовать его взаимодействия с доменами ACT в структуре белка из Chlorobium tepidum. Рассмотрение связывающих остатков показало, что по большей части взаимодействия, вероятно, не являются специфическими, что служит объяснением тому факту, что и другие аминокислоты, такие, как метионин, также могут связываться в этом сайте и регулировать каталитическую активность (Liberles et al. 2005).
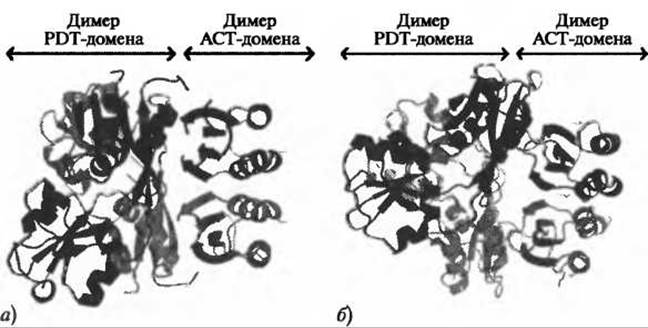
Рис. 11.3. Структуры префенатдегидратазы, полученные в Центре структурной геномики на Среднем Западе, иллюстрируют сходства и отличия: а) структура R-состояния из Staphylococcus aureus (PDB код 2qmw); б) структура Т-состояния из Chlorobium tepidum (PDB код 2qmx). Идентичность последовательностей этих ферментов составляет лишь 27,3%
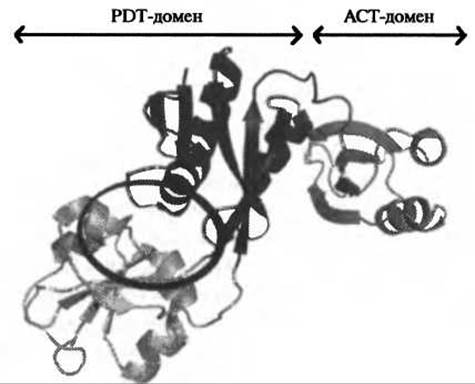
Рис. 11.4. Мономер префенатдегидратазы из Staphylococcus aureus. Каждый домен окрашен в свой цвет. Предполагаемый активный центр расположен в расселине между двумя доменами (обведен)
Сравнение структур префенатдегидратазы из Staphylococcus aureus и Chlorobium tepidum показывает, как связывание L-фенилаланина изменяет конформацию димера ACT. В работе Тан с сотр. (Tan et al. 2008) подробно обсуждается вопрос распространения этих изменений на активный центр, что приводит к блокированию ферментативной активности. Авторы предполагают, что связывание L-фенилаланина вызывает ряд крупных конформационных перестроек в префенатдегидратазе (локальных и глобальных), которые изменяют относительную ориентацию доменов в белке в целом, что приводит к изменению доступности активного центра. Подробнее, эти перестройки приводят к разделению одного широкого просвета, направленного к каталитическому центру в середине димера PDT, на два меньших просвета, что затрудняет доступ префената к каталитическому центру и высвобождение из него фенилпирувата. Этот пример показывает, как анализ структур, полученных в рамках проекта по структурной геномике может иметь значение, выходящее за пределы простого предсказания функции.
Еще одним примером из работ Центра по структурной геномике на Среднем Западе служит белок AF0491 из A. fulgidus (Savchenko et al. 2005), гомологичный белку синдрома Швахмана-Даймонда (СШД) человека. СШД - это редкое аутосомальное рецессивное заболевание, вызванное мутациями в гене SBDS седьмой хромосомы и характеризующееся ненормальной экзокринной функцией поджелудочной железы, дефектами скелета и гематологической дисфункцией (Boocock et al. 2003). Белок AF0491 является архейным гомологом и определение его структуры позволило выявить у него трехдоменное строение (Рис. 11.5).
С-концевой домен имеет широко распространенную укладку, что затрудняет определение его функции. Известно, однако, что такие домены обнаружены во многих РНК- и ДНК-связывающих белках. Центральный домен также имеет распространенную укладку - крыловидный мотив “спираль-поворот-спираль” (winged helix-tum-helix, wHTH). Такой домен часто используется при связывании ДНК (Aravind et al. 2005), а также встречается в РНК-связывающих белках (Schade et al. 1999). В данном случае, однако, поверхность AF0491 не имеет ожидаемого основного характера, поэтому маловероятно, что функция домена состоит в связывании нуклеиновых кислот. Скорее, как это предположили авторы, домен может участвовать в белок-белковых взаимодействиях.
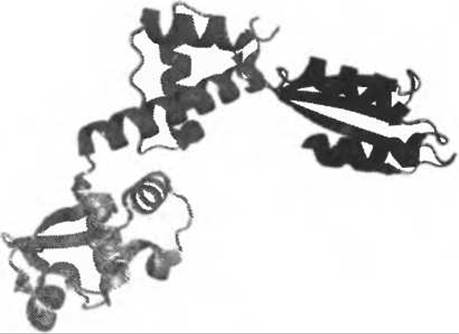
Рис. 11.5. Мономер белка AF0491 из A. fulgidus, гомологичный белку синдрома Швахмана-Даймонда (СШД) человека. Три домена этого белка показаны светло-серым, темно-серым и черным в порядке от N- к С-концу
N-концевой домен имеет новый тип укладки, и именно в этом домене локализована большая часть связанных с заболеванием мутаций, которые выявлены у пациентов с СШД. При последующем структурном поиске этот же тип укладки был найден в дрожжевом белке YHR087W. Обнаружение этого структурного гомолога дает возможность провести дополнительные эксперименты, неосуществимые с человеческим белком. Экспериментальное изучение гомологов белка СШД по структуре и последовательности (YHR087W и YLR022C соответственно) указывает на их связь с метаболизмом РНК. Штаммы с удаленным геном YLR022C оказались нежизнеспособными, но белки, помеченные TAP-тагом, выделялись совместно с многочисленными рибосомальными белками и белками, участвующими в процессинге рРНК. Штаммы с удаленным геном YHR087W оказались жизнеспособными, поэтому такой штамм был скрещен с другими 383 штаммами, у каждого из которых был удален какой-либо ген из тех, что отвечают за белки, участвующие в метаболизме РНК, и в ряде таких комбинация наблюдалась заметная летальность. Это наблюдение и все генетические взаимодействия, выявленные для YHR087W, говорят в пользу участия этого белка в процессинге РНК. Несмотря на то, что эти данные указывают на связь белка СШД с рибосомальным биогенезом, конкретная роль этого белка в метаболизме остается неизвестной, и фундаментальные отличия между рибосомальным биогенезом в бактериях (Lecompte et al. 2002), эукариотах и археях означают, что любые выводы о функции должны внимательно анализироваться. Однако белок СШД является примером, когда определение структурного бактериального гомолога для человеческого белка помогло найти также гомолог и в дрожжах, что оказалось полезным для определениях их функции.