Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Предсказание функции белков на основе их теоретических моделей
Практическое применение
Пластичность остатков каталитического центра
После того, как модель построена, оценена и, возможно, размещена в базе данных, она должна стать инструментом для лучшего понимания взаимосвязей между структурой и функцией белков. Обычно такой анализ выполняется специалистами по биоинформатике, но потенциально использование БД моделей позволит любому исследователю выяснить, что структура модели белка говорит о его функции. Предыдущие главы этой книги продемонстрировали много различных способов, которые позволяют сделать вывод о функции белка, основываясь на его структуре. Ниже будут представлены примеры, иллюстрирующие применение многих из этих методик к структурной информации, полученной методами моделирования ab initio, распознавания фолда и сравнительного моделирования (Главы 1-3, соответственно).
Несмотря на усилия, предпринятые при выполнении проектов по структурной геномике, по охвату всего пространства укладок белков и получению шаблонов для всех ныне существующих семейств, есть примеры, когда критерий близости последовательностей оказывается недостаточным для того, чтобы определить наличие у анализируемого семейства уже описанной функции. Во многих случаях, однако, выводы о структуре белка могут быть сделаны с помощью методов распознавания фолда - как самих по себе, так и в сочетании с моделированием ab initio (Kolinski and Bujnicki 2005), - а затем применены к поиску потенциального активного центра, который позволит предположить возможную функцию белка. Это может быть проиллюстрировано опубликованным анализом (Feder and Bujnicki 2005) семейства последовательностей, объединенных в кластер COG4636 в базе данных кластеров ортологичных групп (англ. Clusters of Orthologous Groups, COG) (Tatusov et al. 2003) и аннотированного как “неохарактеризованный белок, консервативный в цианобактериях”. Детальный анализ консервативности последовательностей внутри семейства COG4636, дополненный предсказанием вторичной структуры, выявил характерную последовательность а-спиралей и ß-тяжей, связанную с консервативным карбоксильным остатком, которая ранее была идентифицирована в надсемействе нуклеаз PD-(D/E)XK (Bujnicki 2003). Это сходство позволяет предположить, что и члены семейства COG4636 могут принадлежать к этому надсемейству (Рис. 12.1а, b).
Однако множественное выравнивание показало, что только “полумотив” PD является почти идеально консервативным, в то время как ключевой остаток лизина во втором полумотиве (D/E)XK отсутствует. Более точно, на его месте в большинстве членов семейства COG4636 расположен гидрофобный остаток лейцина или валина. Этому факту можно предложить три возможных объяснения. Во-первых, семейство COG4636 может вовсе не относиться к надсемейству PD-(D/E)XK. Во-вторых, оно может относиться к этому надсемейству, но утратило остаток в активном центре и вместе с ним каталитическую активность. И в-третьих, роль утраченного лизина мог взять на себя какой-то другой остаток, но выявить его на основе лишь выравнивания последовательностей не представляется возможным. Будет ли тут полезным предсказание структуры и сможет ли оно помочь определить истинную функцию белков семейства COG4636?
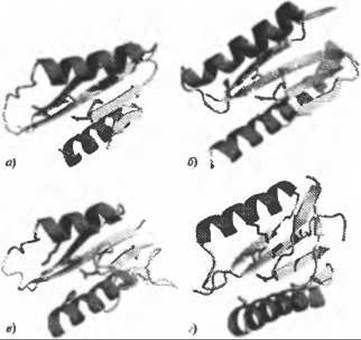
Рис. 12.1. (Цветную версию рисунка см. на вклейке.) Пространственная консервативность активного центра в PD-(D/E)XK. Для наглядности показаны только структурные ядра белков в сходной ориентации, терминальные области и вставки опущены. В верхнем ряду показаны истинные нуклеазы PD-(D/E)XK - а) резольваза структуры Холидея Hje (PDB код 1оb8) и б) рестриктаза Ngo-MIV (PDB код 1fiu).
В нижнем ряду показаны структуры COG4636 - в) теоретическая модель (Feder and Bujnicki 2005) и г) кристаллографическая структура другого члена семейства (PDB код 1wdj). Боковые цепи типичного для PD-(D/E)ХK активного центра и альтерантивного варианта показаны оранжевым (кроме остатков лизина, показанных синим). Этот рисунок, а также все остальные рисунки в данной главе, подготовлен с использованием программы PyMOL (http://pymol.sourceforge.net)
Прежде всего, распознавание фолда для последовательностей семейства COG4636 подтвердило предсказание об их принадлежности к надсемейству ферментов PD-(D/E)XK. Затем по шаблону истинной нуклеазы этого надсемейства была построена сравнительная модель, которую проверили на наличие пространственно близкого консервативного остатка. Оказалось, что в белках семейства COG4636 утраченный лизин замещен другим лизином, заметно удаленным в аминокислотной последовательности (Рис. 12.1с). Функциональная группа этого замещающего лизина может занимать то же пространственное положение, что и группа каталитического лизина в шаблонах, создавая, таким образом, полноценный мотив PD-(D/E)XK в пространстве, несмотря на отсутствие консервативности последовательности. Это наблюдение и позволило сделать нетривиальное предсказание, неосуществимое при анализе одной лишь последовательности, о том, что семейство COG4636 включает в себя активные нуклеазы.
Позднее правильность предсказания необычной конфигурации активного центра была подтверждена кристаллографическими исследованиями другого члена семейства COG4636 (Fig. 12.Id; PDB код lwdj), а также обнаружением других истинных нуклеаз со сходной конформацией активного центра (Tamulaitiene et al. 2006).