Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Распознавание фолда
Определение отдаленной гомологии без протягивания
Использование предсказанных структурных свойств
Одна из самых ранних попыток превратить распознавание гомологии в нечто большее, чем простое сопоставление последовательностей, была предпринята Боуи и сотр. (Bowie et al. 1991). В основе метода лежит тот факт, что определенные структурные свойства белковых последовательностей можно предсказывать в отсутствие точного шаблона. Особенно примечательно, что вторичную структуру, то есть, положение альфа-спиралей и бета-тяжей, теперь можно предсказывать с точностью до 80%, используя такие программы как PSIPRED (Jones 1999а). Учитывая, что структура более консервативна, чем последовательность, пара белков, обладающих отдаленной гомологией, будет содержать схожие паттерны элементов вторичной структуры даже в отсутствие любого очевидного сходства последовательностей. Кроме того, с относительно высокой точностью можно предсказывать степень доступности остатка растворителю (см., например, Kim and Park 2003), а также наличие в структуре крутых поворотов типа бета-шпилька (например, Kumar et al. 2005).
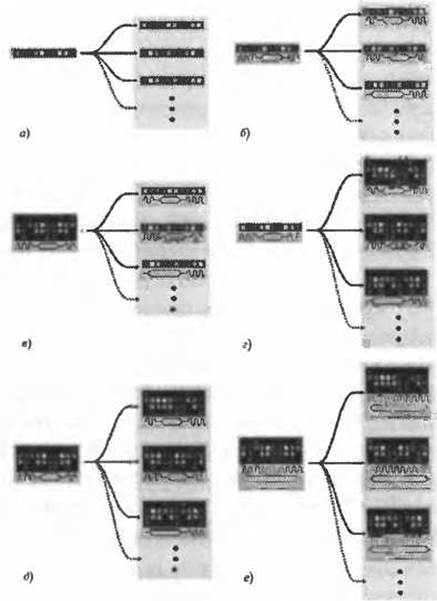
Рис. 2.4. (Цветную версию рисунка см. на вклейке.) Схематическое представление развития методов распознавания фолда на основе последовательностей. В левой части каждого рисунка представлена исследуемая последовательность. Серой ячейкой справа на рисунках показана база данных шаблонов известной структуры. Стрелки указывают на процедуру сравнения исследуемой последовательности с определенным шаблоном, а) Простое сравнение аминокислотной последовательности с последовательностями из базы данных, б) Сопоставление с учетом информации о предсказанной (исследуемый белок) и известной (шаблон) вторичной структуре. Волнистыми линиями показаны альфа-спирали, ромбами - бета-тяжи. в) Исследуемая последовательность представлена профилем из множества последовательностей, PSSM или НММ (СММ) (цветная решетка). Каждому ряду решетки соответствует гомологичная последовательность, каждой колонке - положение в последовательности, г) Ситуация, противоположная (в). Теперь осуществляется поиск исследуемой последовательности в библиотеке профилей. д) Сравнение “профиль-профиль” (вторичная структура по-прежнему представлена простой трехбуквенной строкой), е) То же, что и (д), однако вторичная структура представлена профилем. Для каждого положения последовательности характерны определенные значения вероятности каждого из трех типов вторичной структуры. Обратите внимание, что в данном случае исследователь, вероятно, использует предсказанную вторичную структуру шаблонов, несмотря на то, что фактическая вторичная структура известна. Показано, что метод отличается высокой производительностью, (e.g. Bennett-Lovsey et al. 2008)
Такие предсказанные структурные свойства обеспечивают наличие более подробной информации о структуре белков, которую затем можно использовать наряду с сопоставлением последовательностей. При выравнивании двух аминокислот исследуемой последовательности и последовательности шаблона можно рассчитать совместимость на основе матрицы мутаций, такой как BLOSUM, а также члены, связанные с сопоставлением вторичных структур и доступностью для растворителя:
Sij = Seqij + SSij + Solvij,
где Sij - общая оценка сопоставления остатка і в исследуемой последовательности с остатком j в последовательности шаблона, Seqij - оценка, полученная для сопоставления і и j в матрице BLOSUM, SSij - оценка для сопоставления предсказанного типа вторичной структуры остатка і с известным типом вторичной структуры остатка у, и Solvij — оценка для сопоставления степени заглубленности остатка і с известной степенью заглубленности остатка у. Простые версии таких оценочных функций представлены в таблицах 2.16 и 2.1 в, где одинаковые степени (например, сопоставление спирали со спиралью) получают оценку +1, а все остальные сочетания - оценку -1. Часто функции детально разработаны и основаны на эмпирических наблюдениях частот, с которыми отличающиеся степени оказываются выравнены в случаях известных гомологов. Этот процесс аналогичен продвижению от простой матрицы сопоставления последовательностей на основе идентичности к более чувствительной матрице BLOSUM-типа.
Идея сочетания информации о последовательности и вторичной структуре в ходе поиска по базам данных схематично представлена на рис. 2.46. Методы, в основу которых положена эта идея, значительно превосходят методы стандартного поиска последовательностей по производительности и оказываются на порядки быстрее в вычислительном отношении, чем большинство алгоритмов протягивания. Это свойство является чрезвычайно важным при поиске по большим базам данных шаблонов.
На начальном этапе развития международного соревнования СASP подходы на основе протягивания в целом отличались наиболее высокой производительностью, а описанные выше гибридные подходы “последовательность-структура” вплотную следовали за ними. Однако вскоре последовало смещение методов протягивания с лидирующих позиций, чему способствовали два обстоятельства: 1) стремительное увеличение размеров баз данных последовательностей и 2) развитие метода PSI-BLAST.