Учебник Биология - ВУНМЦ 2000
Глава 4. ОРГАНИЗМЕННЫЙ УРОВЕНЬ ОРГАНИЗАЦИИ ЖИВОГО
4.10. ИЗМЕНЧИВОСТЬ
4.10.2. Наследственная изменчивость
При наследственной изменчивости возникают изменения признаков организма, которые определяются генотипом и сохраняются в ряду поколений. Генотипическая изменчивость может быть комбинативной и мутационной.
4.10.2.1. Комбинативная изменчивость
Комбинативная изменчивость широко распространена в природе. Она является важнейшим источником большого наследственного разнообразия, наблюдаемого у животных организмов. Новые комбинации наследственной информации появляются в результате полового размножения.
Комбинативная изменчивость связана с получением новых сочетаний генов в генотипе, что приводит к появлению организмов с новым фенотипом. Это происходит в результате:
✵ независимого расхождения хромосом при мейозе;
✵ случайного сочетания при оплодотворении;
✵ рекомбинации генов в результате кроссинговера;
✵ взаимодействия генов.
Сами гены при этом не изменяются.
Отличие детей от родителей связано с комбинированием в генотипе детей генов их родителей.
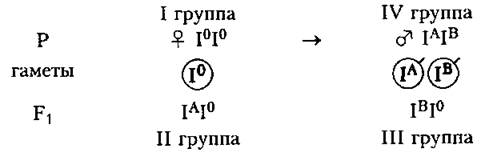
Комбинативной изменчивостью у человека можно объяснить появление у детей II и III групп крови, в отличие от I и IV групп, характерных для их родителей.
Селекционеры часто используют скрещивание отличающихся друг от друга пород и сортов для получения новых. У гибридов, возникших в результате скрещивания, проявились не только новые сочетания признаков, но и новые признаки. Например, при скрещивании кур с розовидным гребнем с породой, обладающей гороховидным гребнем закономерно появились особи с ореховидным гребнем.
С комбинативной изменчивостью связано явление гетерозиса - повышенной гибридной силы - которая наблюдается в 1-м поколении при гибридизации между разными сортами растений. У гибридов увеличивается рост, жизнеспособность, урожайность. Ярко выражен гетерозис у кукурузы.
Гетерозис можно объяснить тем, что:
1. У гибридов увеличивается число доминантных генов, влияющих на развитие признака. Например, если предположить, что на рост влияют гены А и В, то в результате брака представителей с генотипами ААвв и ааВВ ребенок с генотипом АаВв будет иметь более высокий рост:
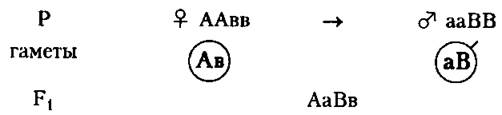
В данном случае имеет место комплементарное действие генов. 2. Иногда гетерозисный организм имеет более выраженные признаки, чем доминантный гомозиготный.
4.10.2.2. Мутационная изменчивость
Мутацией (лат. mutatio - перемена) называют внезапные наследственные изменения генетического материала, возникающие без видимых причин (спонтанно), или могут быть индуцированы внешним воздействием на организм. Процесс возникновения мутаций называют мутагенезом. Факторы, способные вызвать мутации - мутагенами. Организм, приобретший новый признак в результате мутации и изменивший свой фенотип, называют мутантом. Мутации имеют следующие свойства:
✵ они возникают внезапно, скачкообразно;
✵ наследственны, т.е. передаются из поколения в поколение;
✵ ненаправленные - может мутировать любой локус хромосом;
✵ одни и те же мутации могут возникать повторно;
✵ мутации могут быть полезными и вредными, доминантными и рецессивными.
Доминантные мутации проявляются в фенотипе в 1-м поколении. Если доминантные мутации вредные и проявляются и в гомозиготном, и в гетерозиготном организмах, то очень часто организмы оказываются нежизнеспособными и погибают на ранних этапах онтогенеза.
Большинство мутаций рецессивно, не проявляется у гетерозигот и способно накапливаться в генофонде видов, уклоняясь от действия естественного отбора. Мутации часто оказываются вредными, потому что способны нарушать ход биохимических реакций.
При изменении условий внешней среды некоторые ранее вредные рецессивные мутации могут оказаться полезными, и организмы, имеющие их, получат преимущества при естественном отборе.
Мутации, не совместимые с жизнью, называют летальными. Мутации, резко снижающие жизнеспособность, называются полулетальными. Например, ген гемофилии, ген серповидно - клеточной анемии, определяющие синтез аномального гемоглобина.
По месту возникновения мутации бывают генеративными (возникают в половых клетках и проявляются в следующих поколениях) и соматическими (возникают у данного организма, не передаются по наследству при половом размножении и передаются при бесполом).
Соматические мутации возникают часто и остаются незамеченными, но если в некоторых случаях при этом образуются клетки с повышенной скоростью роста и деления, то они могут дать начало опухолям (рис. 171).
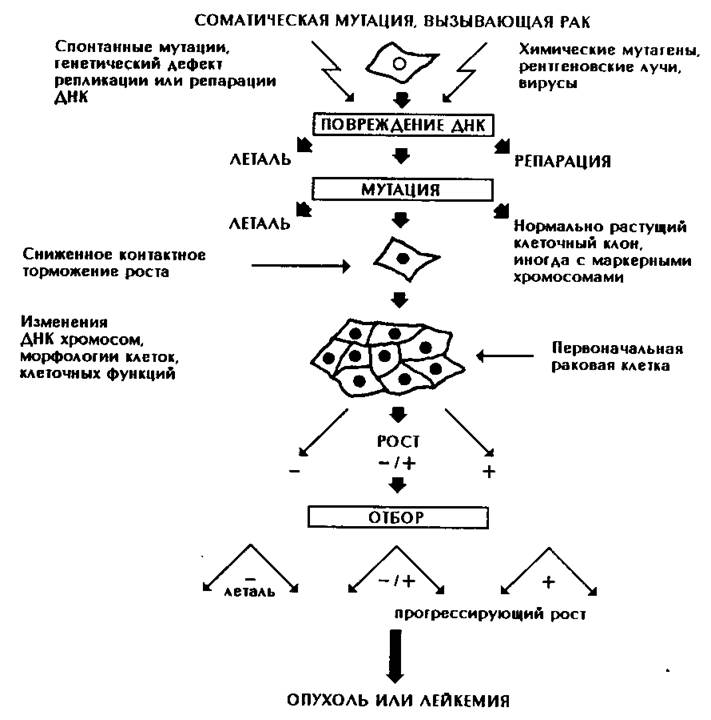
Рис. 171. Развитие злокачественной опухоли. Соматическая мутация, например, хромосомный разрыв, может привести к образованию клона клеток, обладающих селективным преимуществом. Этот клон может постепенно развиться в злокачественную опухоль.
По уровню возникновения мутации могут быть связаны с изменением:
✵ структуры гена - генные;
✵ структуры хромосом - хромосомные перестройки;
✵ числа хромосом (полиплоидия, гетероплоидия) - геномные.
4.10.2.2.1. Генные мутации
Генные мутации образуются наиболее часто и затрагивают структуру гена. Ген - участок молекулы ДНК. Генные мутации возникают при изменении химической структуры гена. Это происходит в результате замены одной или нескольких пар азотистых оснований, или мутаций со сдвигом рамки считывания информации, связанных с выпадением или вставкой одного или нескольких азотистых оснований (рис. 172).
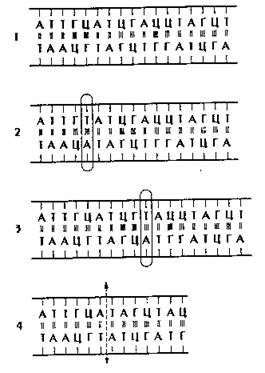
Рис. 172. Главные классы генных мутаций. 1 - нормальная последовательность нуклеотидов, 2 - замена пары "Ц-Г" на пару "А-Т", 3 - вставка пары "Т-А", 4 - потеря блока из шести пар оснований.
Мутации, затрагивающие одну пару оснований и приводящие к замене на другую, удвоению, делеции, называют точковыми. Происходит нарушение последовательности нуклеотидов в молекуле ДНК. Это приводит к изменению строения белка. Генные мутации возникают при замене, выпадении, вставке пар нуклеотидов. Большинство мутаций - генные. С ними связаны изменения морфологических, биохимических, физиологических признаков.
При замене одного пуринового основания на другое или одного пиримидинового на другое возникают транзиции: А⇔Г, Т⇔Ц. Возможны четыре типа транзиции. Транзиции возникают при репликации ДНК.
Могут также меняться пиримидиновые основания на пуриновые и наоборот. Такие замены называют трансверсиями. Их восемь типов: А⇔Т, А⇔Ц, Г⇔Ц, Г⇔Т.
Замены оснований приводят к появлению двух типов мутантных кодонов в и-РНК с измененным смыслом (миссенс - кодон) и бессмысленного (нонсенс-кодон). В результате миссенс-мутации может быть заменена одна аминокислота на другую. Это приводит, например, к появлению аномального гемоглобина при серповидно-клеточной анемии, когда в молекуле гемоглобина глутаминовая кислота заменена валином. В результате такого изменения гемоглобин 5 кристаллизуется при более низкой контцентрации кислорода. В венозной крови эритроциты с таким гемоглобином деформируются, становятся серповидными и быстро разрушаются. У человека развивается анемия. Снижается количество кислорода, переносимого кровью. Люди, гомозиготные по мутантному рецессивному аллелю, быстро погибают. У гетерозигот развивается анемия в слабой форме. Аномальный гемоглобин составляет 40%. Носители аллеля серповидно-клеточной анемии невосприимчивы к малярии.
В результате генных мутаций возникают новые аллели или целые серии мутаций и появляются множественные аллели.
Миссенс-мутации могут влиять на активность ферментов и приводить к синтезу менее активных ферментов или снижать их количество.
Генные мутации способны привести к появлению заболеваний, связанных с нарушением обмена веществ.
Например, заболевание фенилкетонурия возникает при рецессивной генной мутации, приводящей к отсутствию активности фермента фенилаланингидроксилазы. Фенилкетонурия наследуется аутосомно - рецессивно. Ген мутантного фермента находится в одной из аутосом, т.е. болеют и мальчики, и девочки. Заболевание проявляется только у гомозигот, имеющих ген фенилкетонурии в обеих гомологичных хромосомах.
Если родители гетерозиготны по гену фенилкетонурии, то может родиться больной ребенок. Заболевание встречается с частотой 1:7000 родившихся детей.
При фенилкетонурии в результате генетического дефекта фермента происходит накопление в организме большого количества фенилаланина и фенилпировиноградной кислоты. С помощью специальных химикатов у новорожденных детей можно обнаружить в моче фенилпировиноградную кислоту или повышенное содержание фенилаланина в крови.
При фенилкетонурии наблюдаются умственная отсталость и замедленное психическое развитие ребенка, поэтому необходима ранняя диагностика заболевания. Раннее выявление заболевания и исключение из питания продуктов, содержащих фенилаланин, спасают ребенка от тяжелых осложнений.
У человека известно не менее 120 заболеваний, связанных с генными мутациями. Обычно это врожденные дефекты различных ферментов, при участии которых протекают биохимические реакции в организме.
Фенилкетонурия - пример ферментопатии. К заболеваниям, связанным с нарушением обмена аминокислот, относят гистидинемию. При этом заболевании имеется врожденный дефект фермента гистидазы, расщепляющей гистидин. В организме накапливается большое количество гистидина и продуктов его распада.
Это заболевание сопровождается умственной отсталостью, неразборчивой речью, снижением пигментации кожи и волос.
Есть мутации генов ферментов углеводного обмена, приводящие к появлению заболеваний, именуемых гликогенозами. Эти заболевания развиваются в результате генетических дефектов различных ферментов, участвующих в распаде гликогена. При гликогенозах наблюдается избыточное отложение гликогена в скелетной мускулатуре, сердечной мышце или печени. При некоторых формах гликогенозов может развиваться умственная отсталость, мышечная слабость и печеночная недостаточность.
Генные мутации бывают причиной ненормального обмена жиров и жироподобных веществ. Заболевания, именуемые липидозами, сопровождаются тяжелой умственной отсталостью, нарушением функций нервной системы.
Иногда встречаются наследственные дефекты обмена нуклеиновых кислот. При заболевании оротовой ацидурией происходит блокада в системе пиримидиновых нуклеотидов. Мутанты, гомозиготные по данному гену, гибнут внутриутробно, Дети с этим заболеванием умственно отсталы. В их органах и тканях есть отложения оротовой кислоты.
Генетический дефект синтеза пуриновых оснований - причина синдрома Леш - Найхана, который передается сцепленно с полом. Болеют только мальчики. При этом в почках и других тканях накапливается мочевая кислота и возникает подагра.
Нонсенс - мутации приводят к тому, что может появиться нонсенс - кодон не в конце структурного гена, а раньше, что приводит к обрыву полипептидной цепи.
Мутации со сдвигом рамки (фреймшифт), обусловленные вставками или выпадениями одного или нескольких нуклеотидов, напоминают нонсенс - мутации, т. к. приводят к образованию нонсенс - кодонов.
В результате генных мутаций может измениться смысл биологической информации, закодированной в генах. Если условия обитания меняются мало, то возникшие мутации обычно снижают выживаемость вида. Если условия обитания меняются, то наличие мутантных особей может быть полезным.
Появление мутаций связано с нарушением структуры молекулы ДНК. Процесс реконструкции поврежденной ДНК называют восстановлением или репарацией ДНК (рис. 173). Репарация наследственного материала заключается в ферментативном разрушении измененного участка молекулы ДНК с восстановлением на этом участке последовательности нуклеотидов, комплементарной фрагменту неповрежденной молекулы ДНК.
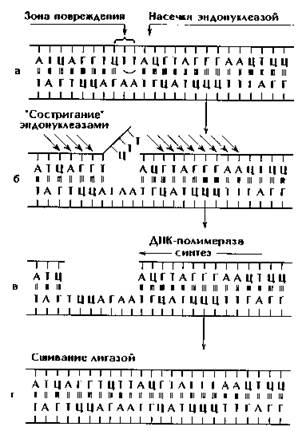
Рис. 173. Этапы репарации ДНК путем вырезания.
В некоторых случаях фермент может разрушить фрагмент нормальной молекулы ДНК, комплементарной измененному, в результате чего образуется мутантная двойная спираль.
Так как молекула ДНК - двойная спираль, то образование генной мутации происходит в два этапа. Сначала изменение затрагивает одну молекулу биоспирали. Это называется молекулярной гетерозиготностью или потенциальной мутацией. Если эти изменения затрагивают гомологичный локус комплементарной молекулы, то возникает истинная мутация и достигается состояние молекулярной гомозиготности. Мутация наследуется всеми потомками мутировавшей клетки. Переход в состояние молекулярной гомозиготности является результатом ошибок репарации.
Репарация или коррекция молекулярных нарушений структуры ДНК приводит к устранению из наследственного материала клетки измененного участка. Различают три основных механизма репарации ДНК (рис. 174).
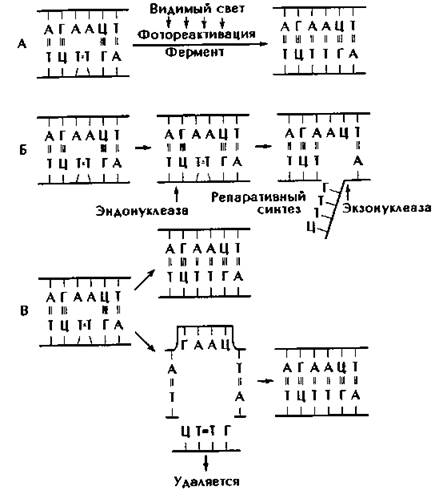
Рис. 174. Три механизма репарации ДНК. А - фотореактивация: тиминовые димеры расщепляются и восстанавливается водородная связь А-Т, Б - эксцизионная репарация: полухроматидная последовательность, содержащая тиминовый димер, вырезается и синтезируется новая полухроматида, В - пострепликативная (рекомбинационная) репарация: полухроматидная последовательность вырезается, репарация происходит после репликации с участием другого продукта деления.
1. Фотореактивация. Действие видимого света на клетки, предварительно обработанные УФ - излучением, приводит к снижению летального эффекта в несколько раз, т.е. к реактивации функций облученных клеток. Реактивирующее действие видимого света связано с расщеплением пиримидиновых димеров. Этот процесс обеспечивается светозависимым фотореактивирущим ферментом.

2. Темновая репарация.
В отличие от фотореактивации в данном случае репарация поврежденной ДНК не нуждается в энергии видимого света. Этот процесс также происходит при участии ферментов. Тиминовые димеры вырезаются из цепи ДНК, в которой остаются бреши. На их места при участии фермента ДНК-полимеразы восстанавливается участок молекулы ДНК, в соответствии с информацией, имеющейся на комплементарной цепи. Фермент ДНК - лигаза принимает участие в восстановлении репарируемой молекулы ДНК.
3. Пострепликационная репарация функционирует в синтетическом периоде митотического цикла. В премитотическом периоде участки молекулы ДНК, имеющие тимидиновые димеры -Т-Т-, не редуплицируются, на их месте образуются бреши. Недостающие фрагменты достраиваются в соответствии с комплементарностью цепи ДНК, что позволяет синтезировать нормальную молекулу ДНК и избежать наследования первичного мутационного изменения дочерними клетками.
4.10.2.2.2. Хромосомные перестройки
Хромосомные перестройки возникают в результате разрыва хромосомы. Перестройки могут быть внутрихромосомными и межхромосомными.
Хромосомные мутации изменяют дозу генов, вызывают перераспределение генов между группами сцепления, меняют локализацию их в группе сцепления.
Внутрихромосомные перестройки, связанные с утратой части хромосомы, называют делениями. Концевые делеции называют дефишенси или нехватки. Они связаны с утратой теломерного участка хромосомы.
![]()
Интерстициальные делеции образуются в результате выпетливания внутреннего участка хромосомы.
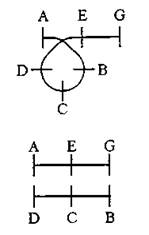
Перестройки, приводящие к удвоению генетического материала, называют дупликациями.
![]()
Дупликациям предшествуют делеции в идентичных участках хромосом. Дупликации могут возникнуть при неравном кроссинговере, если разрывы хромосом происходят не в идентичных участках хромосом, то тогда обмен будет иметь место в неравных участках. В результате такого обмена локус гена в одной из гомологичных хромосом может удваиваться, а в противоположной хромосоме образуется его нехватка.
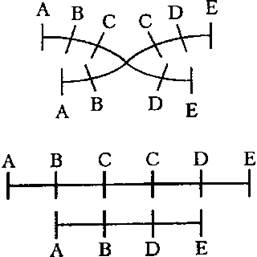
Дупликации и делеции приводят к изменению дозы генов. Перестройки, в основе которых также лежит образование петли с последующим поворотом выпетленного участка на 180° и соответствующим изменением порядка расположения генов, называют инверсией.
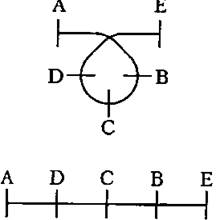
К перестройкам могут быть отнесены также транслокации - перемещения участков на другие места хромосомы или обмен участками между различными хромосомами.
У человека известна делеция 5-й хромосомы. Эта делеция выражается в синдроме "кошачьего крика". Делеция, укорочение на 1/3 короткого плеча 5-й хромосомы, приводит к тому, что у новорожденного имеется много аномалий, умственная отсталость, крик похож на кошачий (рис. 175).
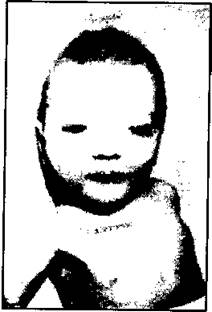
Рис. 175. Больной с синдромом "кошачьего крика" (Macintyre и др., 1964).
Описаны делеции и по другим хромосомам (рис. 176). Их наличие приводит к порокам развития и летальному исходу.
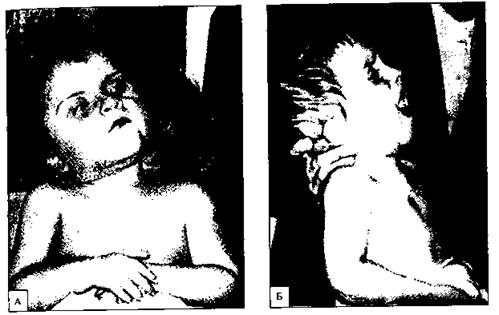
Рис. 176. Ребенок с делецией длинного плеча 18-й хромосомы (Macintyre и др., 1965).
Дупликации могут возникать по всем хромосомам. В результате этого появляются пороки развития, снижающие жизнеспособность организма. Например, дупликация участка 9-й хромосомы может привести к порокам мозговой и лицевой частей черепа и других костей, порокам сердца и различных органов.
В случае инверсии участок хромосомы разворачивается на 180°, и разорванные концы соединяются в новом порядке. Если в инвертированный участок попадает центромера, то такую инверсию называют перицентрической. Если инверсия затрагивает только одно плечо хромосомы, то она называется парацентрической. Гены в инвертированном участке хромосомы располагаются в обратном, по отношению к исходному в хромосоме, порядке.
К межхромосомным перестройкам относят транслокации - обмен сегментами между хромосомами (рис. 177).
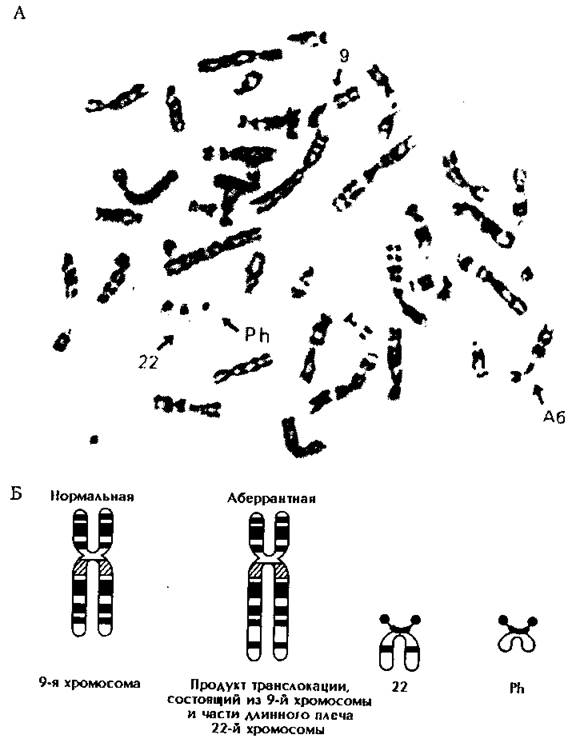
Рис. 177. Филадельфийская (Ph) хромосома у больного с хроническим миелоидным лейкозом.
А - транслокация между 22-й и 9-й хромосомами: Аб - аберрантная хромосома, Б - схематическое изображение исходных хромосом и продуктов транслокации.
Различают несколько типов транслокаций:
✵ реципрокная транслокация, когда две хромосомы взаимно обмениваются сегментами;
✵ нереципрокная транслокация, когда сегменты одной хромосомы переносятся в другую;
✵ транслокация типа центрического соединения, когда после ржрывов в околоцентромерном районе соединяются два фрагмента с центромерами таким образом, что их центромеры соединяются, образуя одну.
Синдром Дауна может быть примером такой транслокации. В кариотипе у больных насчитывается 46 хромосом. Транслокация с 21-й хромосомы на 15-ю (рис. 178).
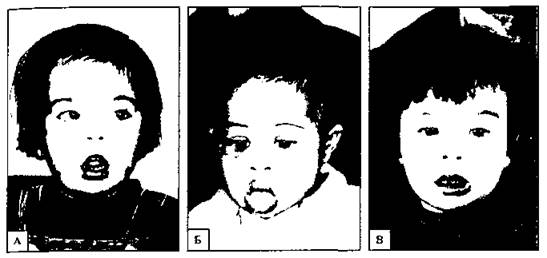
Рис. 178. Дети с синдромом Дауна. А - европеоид, Б - негр, В - представитель азиатской расы. Общие признаки синдрома Дауна более заметны, чем расовые различия (С).
Транслокационная форма характерна для синдрома Эдвардса, но встречается очень редко.
Транслокационная форма характерна и для синдрома Патау, когда в кариотипе больного имеется 46 хромосом. Это происходит чаще всего в результате слияния двух хромосом (13-15). Средний возраст матерей, родивших детей с транслокацией хромосом, не превышает 25 лет.
Внешний вид больных с синдромом Патау специфичен. Больные новорожденные имеют нормальные размеры и массу тела. Клинически отмечается резкая умственная отсталость, выраженная микроцефалия, неправильно сформированные и низко расположенные уши, аномалии глазного яблока, незаращение губы и неба, полидактилия, врожденные пороки сердечно-сосудистой и мочеполовой систем, желудочно-кишечного тракта. Пороки сильно выражены, и дети быстро умирают.
4.10.2.2.3. Геномные мутации
Мутации, связанные с изменением числа хромосом, называют геномными. Совокупность взаимодействующих генов в гаплоидном наборе хромосом клеток организма называют геномом. Геномными мутациями обусловлено появление полиплоидных организмов, когда происходит нарушение кратности полного гаплоидного набора хромосом (триплоидии, тетраплоидии, когда каждая клетка организма содержит не два, а три, четыре гаплоидных набора) или изменение в одной из пар хромосом в сторону утраты гомолога (моносомия) или приобретения дополнительного (трисомия, тетрасомия). В основе численных хромосомных изменений лежат нарушения в расхождении хромосом при клеточном делении. Нерасхождение хромосом может возникнуть во время гаметогенеза, или при первых делениях оплодотворенной яйцеклетки.
К геномным мутациям относят гаплоидию, полиплоидию, анеуплоидию (гетероплоидию). Гаплоидные организмы имеют по одной хромосоме каждой гомологичной пары, все рецессивные гены проявляются в фенотипе. Жизнеспособность организмов снижена.
У человека описаны триплоидные и тетраплоидные организмы. Частота их возникновения низка. Они обнаруживаются среди спонтанно абортированных эмбрионов или плодов и у мертворожденных. Продолжительность жизни новорожденных с такими нарушениями - несколько дней.
Геномные мутации по отдельным хромосомам многочисленны. Моносомии могут быть по Х - хромосоме, что приводит к развитию синдрома Шерешевского- Тернера (45 хромосом = 44 аутосомы + ХО) (рис. 179).
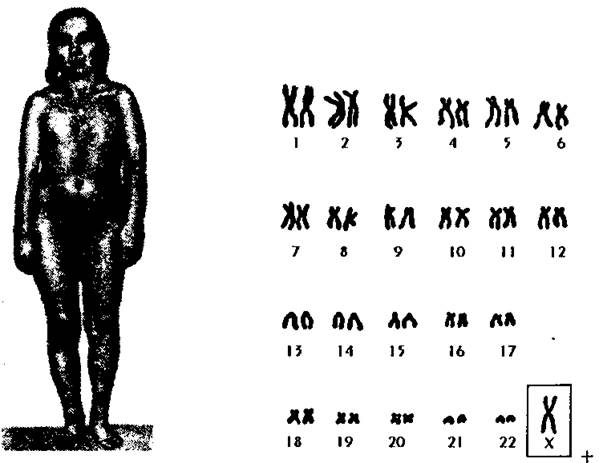
Рис. 179. Моносомия Х (синдром Шерешевского-Тернера) и кариограмма при этом нарушении.
В период созревания гамет наблюдаются случаи нерасхождения половых хромосом (в I, II или в обоих делениях созревания).
Гаметы несут не 22 аутосомы + 1 половую хромосому (X или У), а возникает нарушение парности хромосом. Моносомия Х зависит исключительно от отца.
Для женщин с синдромом Шерешевского-Тернера характерны маленький рост, короткая шея, воронкообразная грудина, бесплодие вследствие недоразвития яичников, слабое развитие половых признаков. 50% больных умственно отсталы или нормальны. Могут быть пороки развития внутренних органов. Дети с синдромом Шерешевского-Тернера рождаются с частотой 0,7 на 1000 новорожденных девочек.
Диагноз ставят при исследовании полового хроматина и на основании результатов цитогенетического анализа.
Аутосомные моносомии среди живорожденных очень редки. Это мозаичные организмы с нормальными клетками. Моносомия касается аутосом 21 и 22. Полные трисомии описаны по большому числу хромосом: 8, 9, 13, 14, 18, 21, 22 и Х. Число Х-хромосом у человека может доходить до 5 с сохранением жизнеспособности (рис. 180, 181).
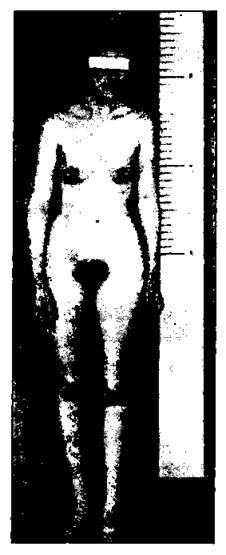
Рис. 180. Фенотип ХХХХ женщины (Сагг и др., 1961).

Рис. 181. Метафазная пластинка и кариотип ХХХХ женщины (Сагг и др., 1961).
Изменение числа хромосом вызвано нарушением распределения их по дочерним клеткам во время 1-го или 2-го мейотического деления в гаметогенезе или при первых дроблениях оплодотворенной яйцеклетки.
Нарушения возникают:
✵ при расхождении во время анафазы редуплицированной хромосомы, в результате чего удвоенная хромосома попадает только в одну дочернюю клетку;
✵ при нарушении конъюгации гомологичных хромосом, что может нарушить правильность расхождения гомологов по дочерним клеткам;
✵ при отставании хромосом в анафазе при их расхождении в дочерние клетки, что может привести к утрате хромосомы (рис. 182).
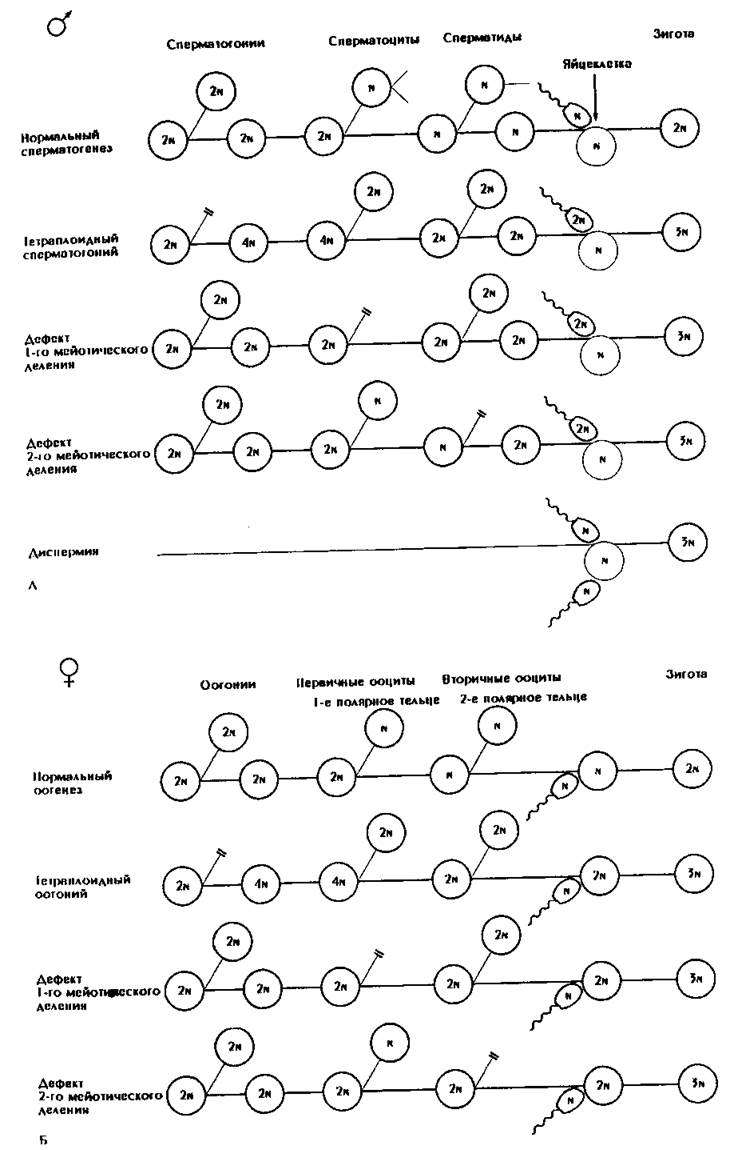
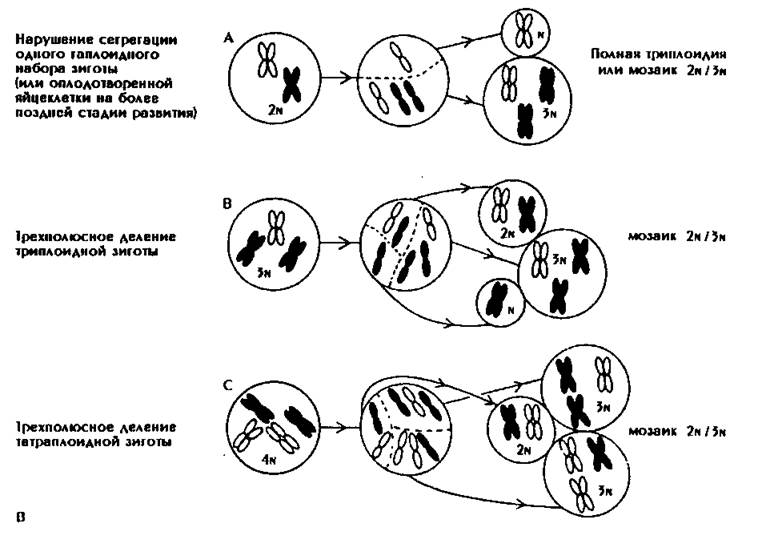
Рис. 182. Аномалии овогенеза, сперматогенеза или оплодотворения, обусловливающие триплоидию.
А - мужчина может оказаться триплоидным, если у его отца имелся тетраплоидный сперматогоиий или был нарушен мейоз. Триплоидия у мужчин может быть и результатом оплодотворения двумя сперматозоидами (по Niebuhr Hum. Genet., 21, 1974). Б - у женщин, так же как и у мужчин, триплоидия может объясняться нарушением гаметогенеза в предыдущем поколении. В - аномальное деление зиготы или клеток эмбриона приводит к мозаицизму.
При нарушении в двух и более последовательных делениях возникают тетрасомии и другие полисемии.
Полисемии по половым хромосомам весьма разнообразны (рис. 183).
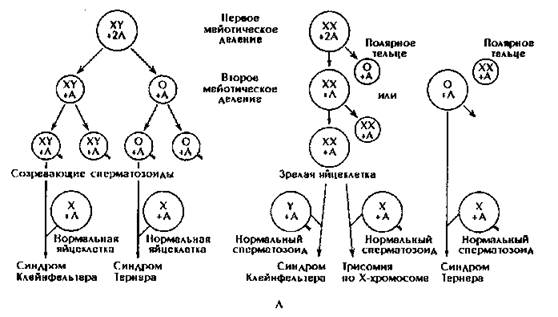
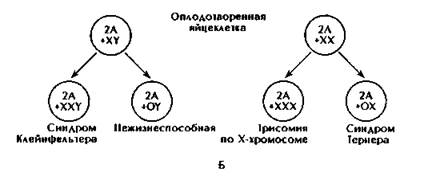
Рис. 183. Возможные механизмы возникновения анеуплоидии половых хромосом.
А - нерасхождение в гаметогенезе. Б - перасхождение или потеря хромосомы па стадии зиготы: показан механизм возникновения немозаичного синдрома Клайнфельтера (тип ХХY) из ХY-зиготы.
Женщины с кариотипом XXX встречаются с частотой 1-1,4 на 1000 родившихся девочек. Для больных с кариотипом XXX характерно наличие недоразвитых яичников, матки, бесплодие. Умственное развитие нормальное или в пределах нижней границы нормы. Около 30% женщин сохраняют способность иметь детей (рис. 184).

Рис. 184. Трисомия Х и кариограмма при этом нарушении.
С увеличением числа Х-хромосом в кариотипе до 4, 5 и более клинические проявления синдрома увеличиваются. Больные не могут иметь детей, умственно более отсталы. При исследовании полового хроматина в ядрах клеток эпителия слизистой оболочки щеки обнаруживают 2 и более телец Барра (рис. 185). Впервые синдром трисомии по Х-хромосоме описали П. Джекобе и др. в 1959 г.
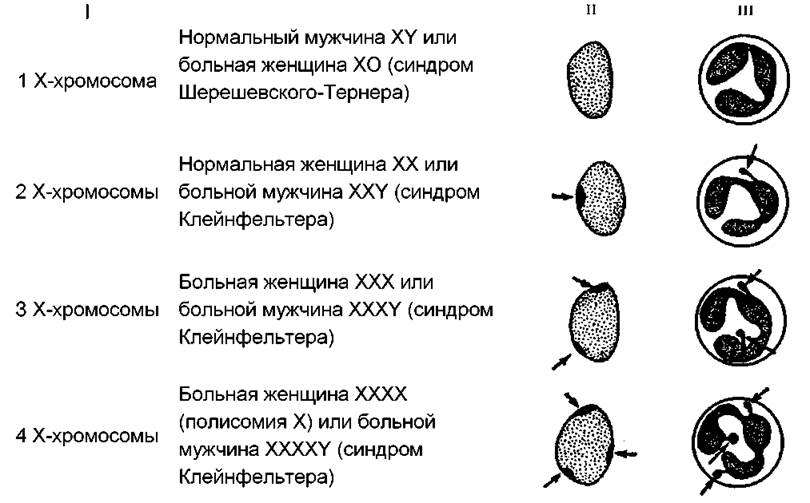
Рис. 185. Связь между числом Х-хромосом (I), числом телец Барра в клетках слизистой оболочки ротовой полости (II) и числом "барабанных палочек" в ядрах лимфоцитов (III).
При синдроме Клайнфельтера, описанном им в 1942 г., у мужчин в ядрах клеток эпителия слизистой оболочки полости рта обнаружено тельце Барра. В кариотипе 47 хромосом (44+ХХY). Частота больных с синдромом Клайнфельтера колеблется в пределах 2-2,5 на 1000 новорожденных мальчиков (рис. 186).

Рис. 186. Синдром Клайнфельтера.
А - клинические признаки: наружные половые органы сформированы по мужскому типу, но яички недоразвиты, оволосение слабо выражено, в большинстве случаев наблюдается гинекомастия, очень длинные ноги, имеется половой хроматин. Б - анализ кариотипа данного больного показал, что у него 47 хромосом, в том числе три половые хромосомы - тип ХХY.
Для мужчин с синдромом Клайнфельтера характерен высокий рост, длинные конечности, евнухоидизм, нарушенный сперматогенез и бесплодие, гинекомастия, повышенное выделение женских гормонов, склонность к ожирению. Лишняя хромосома Х обусловливает разнообразные нарушения психики, снижение интеллекта. Иногда наблюдается антисоциальное поведение и алкоголизм. Степень тяжести симптомов пропорциональна числу добавочных Х-хромосом.
Разновидностью синдрома Клайнфельтера является полисомия по хромосоме Y - синдром ХYY (47 хромосом). У мужчин с хромосомным набором ХYY рост выше среднего, умственное развитие ниже нормы. Они отличаются агрессивным поведением, наблюдается бесплодие. Среди новорожденных мальчики с данным синдромом рождаются с частотой 1:1000.
Индивиды с полисомией по Х- и Y-хромосомам (48-ХХYY, 49-ХХХYY) очень редки - 1:25000 новорожденных мальчиков. Они отличаются снижением интеллекта, агрессивностью поведения (рис. 187).
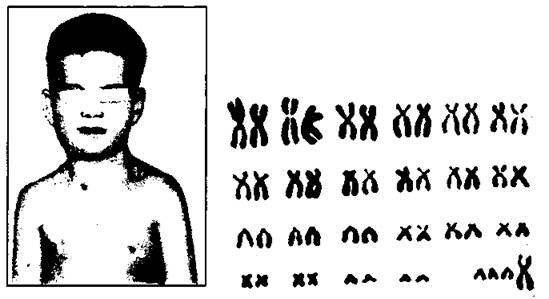
Рис. 187. Фенотип ХYYY при синдроме Клайнфельтера и кариограмма при этом нарушении (Townes и др., 1965).
Полные трисомии описаны по большому числу аутосом: 8, 9, 13, 14, 18, 21,22.
Трисомия по хромосоме 8 приводит к живорождению, но часто наблюдается мозаицизм. Рождение детей с этим геномным нарушением происходит с частотой 1:50000 новорожденных. При синдроме отмечается неглубокая умственная отсталость и физическое недоразвитие. Типичны скелетные аномалии, удлиненное туловище, нарушения речи.
Трисомия по 9-й паре хромосом заканчивается внутриутробной гибелью носителя лишней хромосомы. Продолжительность жизни немногих рожденных детей с такой трисомией-9 составляла 3,5 месяца. Для них характерны внутриутробное недоразвитие, черепно-лицевые пороки, аномалии скелета, пороки сердца, почек и других органов.
Трисомия по 13-й паре хромосом (синдром Патау) - была описана в 1960 г. - встречается с частотой 1:5000-7000 рождений (рис. 188).

Рис. 188. Трисомия D (синдром Патау) и кариограмма при этом нарушении.
Для синдрома характерны пороки головного мозга, лица, внутренних органов (сердца, почек, половых органов), полидактилия. Глухота наблюдается в 80-85% случаев. Имеет место ранняя смертность (в течение первого года погибает 90% детей с синдромом Патау) (рис. 189).
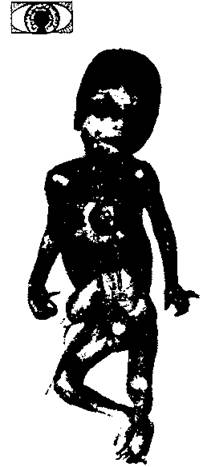
Рис. 189. Основные клинические симптомы трисомии по хромосоме 13 (синдром Патау): колобома - микрофтальм, умственная отсталость, задержка роста, низко расположенные и деформированные уши, глухота, обезьянья складка, дистальный осевой трирадиус, дефект перегородки предсердия, дефект межжелудочковой перегородки, стенокардия, U-образные фибулярные радиальные дуги, увеличенная сегментация полиморфноядерных гранулоцитов, высокая частота "барабанных палочек", микроцефалия, арриненцефалия, гипертелоризм, незаращение верхней губы и неба, полидактилия, флексия, деформация пальцев и ногтей, почечные кисты, двойной мочеточник, гидронефроз, гидроуретер, пупочная грыжа, аномалии развития матки, крипторхизм.
Трисомии по 14-й паре хромосом описаны для мертворожденных. У живорожденных этой патологии не выявлено.
Трисомии по 18-й паре (сидром Эдвардса) встречаются с частотой 1:7000 среди живых младенцев. Для детей характерно пренатальное недоразвитие, пороки костной системы, пороки сердца, отклонения в дерматоглифическом рисунке. 90% детей умирают на первом году жизни (рис. 190, 191).

Рис. 190. Основные клинические симптомы трисомии по хромосоме 18 (синдром Эдвардса): задержка роста, умственная отсталость, долихоцефалия с выступающим затылком, ретрофлексия головы, дуги на трех или более концах пальцев, отсутствие кожных складок выше дистальных суставов, обезьянья складка, короткая грудина, подковообразная почка, аддукционная деформация бедра, мышечный гипертонус, pesequinouarus, выступающие пятки, дорзальная флексия больших пальцев, открытые швы черепа и широкие роднички при рождении, гипертелоризм, высокие надбровные дуги, низко расположенные и деформированные уши, микрогнатия, флексорная деформация пальцев, персистирующий артериальный проток, дефект межжелудочковой перегородки, Меккелев дивертикул, отсутствие больших губ, выступающие наружные гениталии, маленькая плацента.

Рис. 191. Трисомия Е (синдром Эдвардса) и кариограмма при этом нарушении.
Наиболее часто встречается трисомия по 21-й паре хромосом (синдром Дауна). Клиническое описание этого синдрома было сделано в 1866 г. английским врачом Дауном. Мальчики и девочки заболевают одинаково часто. Частота рождения детей с синдромом Дауна - 1:700-800 новорожденных. В большинстве случаев при трисомии в кариотипе 47 хромосом.
Известно, что чем старше мать, тем больше риск рождения ребенка с синдромом Дауна. У матерей старше 40-44 лет риск появления такого ребенка в 16 раз выше, чем у матерей в возрасте 20-24 года, 95% случаев синдрома имеют аутотрисомный вариант.
Больные с синдромом Дауна небольшого роста, слабоумны, имеют физические пороки (рис. 192). Для них характерны небольшая голова со скошенным затылком, косые глазные щели, эпикант, короткий нос с широкой переносицей, маленькие деформированные уши, полуоткрытый рот с высунутым языком и выступающей нижней челюстью, походка с неловкими движениями, косноязычие. Они имеют пороки сердца, желудочно-кишечного тракта, почек. У больных часто возникают инфекционные и злокачественные заболевания, что обусловлено дефектами иммунной системы. Особенности дерматоглифики связаны с глубокой поперечной бороздой (обезьянья складка) и единственной сгибательной складкой на мизинцах (рис. 193). Благодаря улучшению условий жизни и медицинской помощи, больные с синдромом Дауна доживают до 30 лет и более. Некоторые больные могут заниматься посильной трудовой деятельностью.
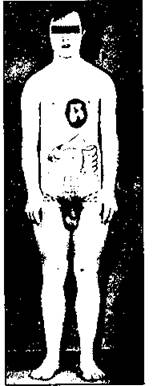
Рис. 192. Основные клинические симптомы болезни Дауна: задержка роста, умственная отсталость, плоский затылок, диснластичные уши, много "петель" на кончиках пальцев, обезьянья складка, срединный осевой трирадиус, одностороннее или двустороннее отсутствие одного ребра, стеноз кишечника, пупочная грыжа, диспластичный таз, гипотоничные мышцы, широко отставленные большие пальцы, широкое плоское лицо, раскосые глаза, эпикант, короткий нос, маленькое и арковидное небо, большой складчатый язык, зубные аномалии, короткие и широкие кисти, клинодактилия, врожденный порок сердца, мегаколон.
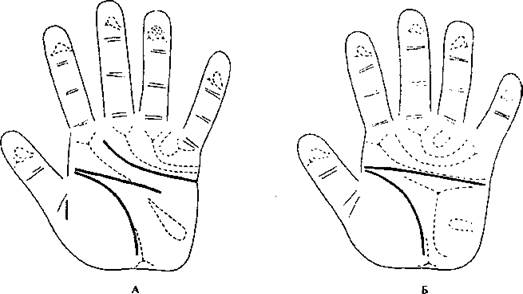
Рис. 193. Дерматоглифика при болезни Дауна.
А - ладонь нормального субъекта, Б - ладонь при болезни Дауна (Turpin, Lejeune, 1965).
Трисомия по 22-й паре, как правило, вызывает летальный эффект и гибель плода во внутриутробном периоде.