БИОЛОГИЯ Том 3 - руководство по общей биологии - 2004
25. ПРИКЛАДНАЯ ГЕНЕТИКА
25.7. Генетика человека
25.7.4. Фенилкетонурия
Фенилкетонурией заболевает примерно один из 10 000 новорожденных белых, а каждый из 80таких младенцев является носителем. У представителей других рас это заболевание встречается реже.
Причина
Причиной фснилкетонурии, как и муковисцидоза служит рецессивная аутосомная мутация. Это очень тяжелое заболевание, однако ранняя диагностика и лечение могут предотвратить его разрушительное действие на здоровье человека. У больных фенилкетонурией аминокислота фенилаланин не превращается в другую аминокислоту — тирозин:
![]()
У здоровых людей в клетках печени присутствует фермент фенилаланингидроксилаза, однако у больных фенилкетонурией этого фермента нет. Ген, кодирующий его, находится на хромосоме 12. В результате мутации этого гена в организме больного накапливается фенилаланин. Избыток фенилаланина оказывает губительное действие на мозг человека и его умственное развитие. Новорожденные с фенилкетонурией выглядят вполне нормальными, так как в период внутриутробного развития избыток фенилаланина проходит через плаценту и утилизируется в печени матери. Если лечения не проводить с младенческого возраста, уже в первые годы жизни патология становится заметной. Наиболее серьезный симптом — сильное отставание в умственном развитии. У пациентов, не подвергающихся лечению, коэффициент умственного развития (IQ) может быть меньше 20. До того как были разработаны методы лечения, более 1 % пациентов психиатрических клиник составляли именно больные фенилкетонурией. Они редко доживали до 30 лет. Помимо умственной отсталости среди других симптомов болезни отмечены;
1) гиперактивность у детей;
2) неустойчивая походка;
3) более светлые волосы, кожа и глаза, чем у здоровых родственников (в отсутствие тирозина не синтезируется коричневый пигмент кожи — меланин);
4) кожные изменения, напоминающие младенческую экзему;
5) эпилептические припадки.
25.2. Разберите приведенный ниже пример наследования фенилкетонурии.
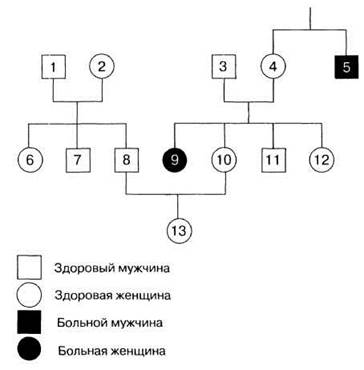
а) Как доказать, что фенилкетонурия контролируется рецессивным геном?
б) Как доказать, что фенилкетонурия не сцеплена с полом?
в) Основываясь на имеющихся данных, скажите, кто из членов семьи является гетерозиготным носителем?
г) Кто из других членов семьи может быть носителем?
д) Индивидуумы под номерами 10, 11 и 12 могут испытывать волнение по поводу предполагаемого носительства, поскольку их сестра больна фенилкетонурией. Если один из них спросит вас, какова вероятность этого события, что вы ответите? Хорошо подумайте!
Выявление фенилкетонурии у новорожденных
Очень важно, чтобы фенилкетонурия была выявлена в первые дни жизни, поскольку в первые шесть месяцев несмотря на отсутствие симптомов у нелеченых детей развиваются необратимые повреждения мозга. В 1963 г. был разработан очень чувствительный метод анализа крови, который позволяет обнаружить в крови больных избыток фенилаланина (обычно это 30—50-кратное превышение). Тестирование проводят на 4-й день жизни; пробу крови для анализа берут из пятки: младенца.
25.3. Почему младенцев не тестируют сразу после рождения?
Выявление носителей и пренатальная диагностика
Современные методы анализа ДНК позволяют проводить скрининг популяции. Благодаря этому выявляется до 95% носителей фенилкетонурии. В настоящее время появилась возможность пренатальной диагностики фенилкетонурии; для этого берут пробы клеток из ворсинок хориона или амниотической жидкости и затем проводят анализ ДНК. Однако целесообразность этой процедуры неочевидна, поскольку в утробе плод не страдает от избытка фенилаланина, а лечение начатое с первых дней жизни, предупреждает развитие симптомов. Вероятно, в будущем станет возможной генная терапия.
Лечение
С заболеванием борются путем снижения количества фенилаланина в рационе больных до необходимого минимума. Фенилаланин является незаменимой аминокислотой (это означает, что он не может быть получен из других аминокислот, и, следовательно, для нормального синтеза белка должен присутствовать в рационе). Поскольку в организме больных не происходит образования тирозина из фенилаланина, тирозин также является для них незаменимой аминокислотой и тоже должен присутствовать в пище. Для поддержания необходимого баланса в первые годы жизни осуществляют постоянный контроль содержания этих аминокислот в крови больного. В зрелом возрасте, когда уже завершено развитие мозга, избыток фенилаланина не опасен и больным можно переходить на нормальную пищу. Соблюдать диету непросто, поскольку она исключает хлеб, сладости, апельсиновый сок, т. е. все то, что любят дети, однако нарушение диеты приводит к необратимым последствиям.
25.4. Почему больным фенилкетонурией повезло, что фенилаланин является незаменимой аминокислотой?
25.5. Взрослые люди, болеющие фенилкетонурией, уже не обязаны придерживаться диеты. Почему больным женщинам, планирующим беременность, важно вернуться к рациону с ограниченным количеством фенилаланина?