БОТАНИКА ТОМ 2 - ФИЗИОЛОГИЯ РАСТЕНИЙ - 2007
6. ФИЗИОЛОГИЯ ОБМЕНА ВЕЩЕСТВ
Жизненные процессы связаны с постоянным превращением вещества и энергии. Живые организмы поглощают определенные вещества и энергию из окружающей среды и отдают другие вещества и энергию (в особенности тепло) в окружающую среду. Такие системы в термодинамике (греч. therme — тепло; dynamis — движущая сила) называются открытыми системами. В конечном счете энергия, которая привносится в биосферу, в подавляющем большинстве происходит из солнечного света, и в процессе фотосинтеза переводится зелеными растениями в химическую форму. При этом из неорганических веществ образуются органические соединения. Организмы, которые создают из неорганических соединений все необходимые органические вещества, называют автотрофными (первичные продуценты). Если растения используют энергию света, они фотоавтотрофны.Некоторые микроорганизмы живут хемоавтотрофно, т. е. используют как материю, так и энергию неорганических соединений. Гетеротрофные организмы (консументы)получают вещества и энергию от первичных продуцентов. Они таким образом зависят от органических соединений, которые синтезируют первичные продуценты, и покрывают свою потребность в энергии также из поглощаемого органического вещества. Из числа гетеротрофов, сапрофиты1 питаются за счет уже неживых источников питания, паразиты — за счет живущих организмов (табл. 6.1; см. 9.1.1).
1 Среди сосудистых растений сапрофитная стратегия питания в чистом виде не встречается. Как правило, при использовании неживых источников органики сосудистые растения вступают в симбиоз или паразитируют на грибах. — Примеч. ред.
Превращение вещества и энергии в клетке, метаболизм (греч. metabole — изменение, перемена), может быть подразделен на анаболические (создающие) и катаболические (разлагающие) процессы. Основополагающе важные для жизненных функций метаболические пути формируют первичный обмен веществ. Именно растения отличаются в особенности богато дифференцированным вторичным метаболизмом. Ко вторичному метаболизму относят специальные метаболические пути, которые начинаются от метаболитов первичного обмена веществ (и лишь только на этом основании, а ни в коем случае не по их значению, называются вторичными) и ведут к продуктам с дополнительными функциями, часто экологическими, как, к примеру, вещества защиты от поедания животными. Вторичные метаболиты в основном ограничены определенными группами растений и таким образом имеют таксономическое значение.
Таблица 6.1. Различные пути ассимиляции углерода у организмов
Тип питания |
Автотрофия |
Гетеротрофия |
||||
Фотогидротрофия |
Фотолитотрофия |
Хемолитотрофия |
Фотоорганотрофия |
Сапрофитизм |
Паразитизм |
|
Источник энергии |
Свет |
Свет |
Окисление |
Свет |
Диссимиляция |
Диссимиляция |
Источник углерода |
СО2 |
СО2 |
СО2 |
СО2 или органические вещества |
Органические вещества (от уже неживых источников) |
Органические вещества (от живых организмов) |
Донор электронов |
Н2О |
Неорганические вещества (например, Н2S) |
Неорганические вещества (например, Н2S, NН3, Fе2+, Н2) |
Органические вещества |
Если необходимо, диссимиляция |
Если необходимо, диссимиляция |
Встречаемость |
Зеленые растения, цианобактерии, прохлоробактерии |
Сернопурпурные бактерии (Сhromatiа- сеае), зеленые серобактерии (Chlorobiасеае) |
Некоторые бесцветные прокариоты |
Пурпурные бактерии (Rhodospirillасеае), бессерные зеленые бактерия (Chloroflexaсеае) |
Бактерии, грибы, животные |
Бактерии, грибы, некоторые покрытосеменные и красные водоросли, животные |
В первом разделе этой главы вначале будут рассмотрены основные термодинамические принципы жизненных процессов (см.
6.1), затем автотрофные функции растения, начиная с поглощения и переработки минеральных веществ (6.2), которые тесно связаны с водным обменом (6.3). Синтез органических соединений из поглощенных неорганических предшественников с использованием энергии света (фотосинтез) и распределение продуктов фотосинтеза (ассимилятов) в растении составляют два раздела (6.4, 6.5) описания первичного обмена веществ растения (6.4—6.15), за которым следует обзор важных аспектов вторичного метаболизма (6.16) и метаболизма характерных для растения полимеров (6.17). Глава завершается кратким описанием выделительных процессов у растений (6.18).
6.1. Энергетика обмена веществ
Нет сомнений, что превращения вещества и энергии в живых организмах следуют законам физики и химии, а принципы термодинамики, т. е. учения об изменениях энергии при протекании физических или химических процессов, действительны также для живых существ. Если превращения энергии в живой клетке часто объединяют под термином биоэнергетика (греч. еnergia — действие), то это всего лишь означает, что в рамках термодинамически возможных процессов и превращений определенные из них особенно характерны для живой клетки и что задействованные в реакциях типы молекул, прежде всего катализаторы, отличаются от таковых неживой природы и техники.
Живые существа являются открытыми системами (в термодинамическом смысле), т.е. находятся в постоянном обмене энергией и веществом с окружающей их средой. Они развиваются, а это значит, что их материальный и энергетический обмен подвержен изменениям во времени. К тому же жизненные процессы необратимы, и живой организм далеко отстоит от состояния термодинамического равновесия. Живые существа поэтому следует описывать на основе закономерностей необратимой термодинамики неравновесных состояний; что пока (если учесть чудовищную сложность жизненных процессов) представляется не реальным предприятием1. Существенная принципиальная информация может быть получена уже из намного более простой термодинамики равновесных состояний в закрытых системах — системах, которые обмениваются со средой энергией, но не материей. По этой информации можно судить, реальна ли при данных условиях определенная химическая реакция. Законы равновесной термодинамики, однако, ничего не говорят о том, как быстро реакция будет протекать.
1 Полное описание всех термодинамических процессов в клетке невозможно в принципе, поскольку она связана с внешней средой. Всегда можно добавить новый фактор среды, который изменит метаболизм клетки, и тогда описание окажется неполным. — Примеч. ред.
Метаболизм живой клетки служит для того, чтобы выполнять определенные функции и совершать работу, для которой требуется энергия.
Абсолютной мерой работы (сила х путь), как и энергии, является Джоуль (Дж; 1 кг 1м2 1 с-2 = кг м2 с-2), т.е. единица силы (ньютон, Н: кг м С-2) х единица пути (м). Часто можно встретить данные в килоджоулях (кДж; 103 Дж). Ранее используемой и еще часто встречающейся мерой энергии является калория (1 кал = -4,184 Дж), мера теплоты (1 кал соответствует количеству энергии, необходимому для повышения температуры 1 г воды при нормальном давлении с 14,5 до 15,5 °С). Правомочность применения этой единицы как всеобщей меры энергии обусловлена взаимообратимостью отдельных форм энергии, переходом одной в другую (например, кинетической, тепловой, химической, электрической и энергии излучения). Предпочтение для единицы теплоты было основано на том, что тепло есть наиболее общая форма энергии, все остальные виды энергии могут быть без остатка переведены в тепло (но не наоборот). Для температуры чаще используется единица в градусах Цельсия (°С), хотя корректно использовать абсолютную температуру в кельвинах, К (0 К = -273,15 °С). (Таблица с единицами СИ и пересчетными коэффициентами находится в конце книги.)
6.1.2. Энергетика закрытых систем
Термодинамика рассматривает, как правило, поведение (точнее, изменение состояния, А) некой ограниченной области (системы). Все, что находится вне системы, есть ее окружающая среда. Система и окружающая среда называются «целым», «вселенной», «universum» (рис. 6.1). Система обладает внутренней энергиейU, которая представляет сумму всех форм энергии в системе. Первый закон термодинамики гласит, что внутренняя энергия замкнутой системы, т. е. системы, которая не обменивается со средой ни материей, ни энергией, постоянна (U = const). Количество энергии в системе зависит от состояния системы, но не от того, каким образом это состояние было достигнуто. Поэтому для кругового процесса, при котором система вновь возвращается в свое начальное состояние, ∆U = 0. Таким образом, энергия не может быть создана из ничего и не может быть потеряна.
Рис. 6.1. Определения различных термодинамических систем
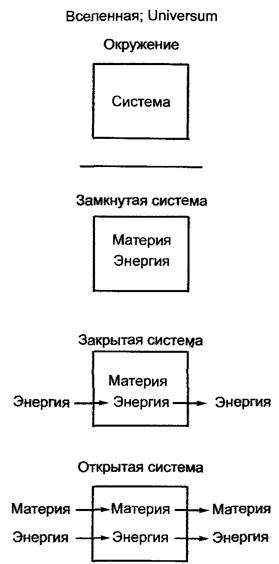
Если извне в систему вносится энергия, например, определенное количество теплоты (Q) (тогда по определению это не замкнутая, а закрытая система, так как она обменивается с окружающей средой энергией, но не материей), то в соответствии с первым законом привнесенное тепло ведет или к изменению внутренней энергии системы, или к производству работы (W):
Q = ∆U + W или ∆U = Q - W. (6.1)
Процессы, при которых система поглощает тепло, называются эндотермическими, процессы, происходящие с выделением тепла, — экзотермическими. В реакциях при постоянном давлении (р = const), как они в общем случае происходят в организмах, изменение теплоты называется изменением энтальпии и обозначается ∆Н (Q = ∆Н). Тогда
∆U = ∆Н - W, (6.2)
причем W в общем случае осуществляется как работа по изменению объема: W = p∆V. При постоянном объеме и постоянном давлении, таким образом, не производится никакой работы (W = 0), и
∆U = ∆Н.
При этих условиях через определение теплоты реакции можно сделать заключение об изменении энергии во время ее протекания. Изменение энтальпии (∆Н) реакции может быть определено путем калориметрии (греч. calor — тепло; métrein — мерить). Если ∆Н > 0, говорят об эндотермическом, а если ∆Н < 0, — об экзотермическом процессе. Органические соединения имеют определенную молярную теплоту сгорания, которая выражается как энергия (Дж), высвобождающаяся в окружающую среду при полном окислении 1 моля вещества (табл. 6.2).
Таблица 6.2. Теплота сгорания различных органических соединений, важных для обмена веществ клетки
Вещество |
Молекулярная масса, Да |
∆H |
|
кДж/моль |
кДж/г |
||
Глюкоза С6Н12O6 |
180 |
-2 817 |
-15,65 |
Молочная кислота CH3-CHOH-COOH |
90 |
-1364 |
-15,16 |
Щавелевая кислота HOOC-COOH |
90 |
-251 |
-2,79 |
Пальмитиновая кислота CH3-(CH2)14-COOH |
256 |
-10037 |
-39,21 |
Трипальмитин C51H98O6 |
809 |
-31433 |
-39,00 |
Глицин NH2CH2-COOH |
75 |
-979 |
-13,05 |
Первый закон термодинамики не позволяет сделать вывод о направлении протекания физических или химических процессов. Из общего опыта, однако, известно, что самопроизвольно (т.е. автономно) протекающие процессы имеют направление. Так, например, тепло переходит от более теплого к более холодному телу; обратный процесс еще никогда не наблюдался. B общем случае известно, что лишь состояния с более низкой организацией спонтанно возникают из состояний с более высокой организацией, причем систему и ее окружение нужно рассматривать в совокупности. Мерой беспорядка служит термодинамическая функция S, энтропия (греч. entrepein — превращать). Каждое самопроизвольное изменение состояния связано с увеличением энтропии. Это — одна из формулировок второго закона термодинамики. Белковая молекула, самопроизвольно переходящая из развернутой конформации с низкой степенью организации в свернутое, более высоко организованное состояние при формировании вторичной и третичной структур, кажется противоречащей этой закономерности. Процесс свертывания, однако, протекает при нарушении структуры воды в окружении свертывающейся молекулы белка, так что общая энтропия в системе (белок) и в окружающей среде (водная среда) во время свертывания увеличивается. Таким же образом поддержание состояния высокой организации (низкой энтропии) живых существ неизбежно связано с возрастанием энтропии в окружающей их среде.
Размерность энтропии [Дж К-1]. При каждой данной температуре твердые тела имеют относительно низкую энтропию, жидкости — среднюю, газы — высокую. Энтропия возрастает с температурой, так как тогда молекулы имеют большее тепловое движение. Энтропия идеального кристаллического тела в точке абсолютного нуля (-273,15 °С = 0 К) равна нулю (это обстоятельство часто обозначают как третий закон термодинамики).
Как уже упоминалось, приток тепла в систему может быть использован для работы, что, например, происходит в тепловых машинах. В живой клетке, однако, температура практически не изменяется1: клетка функционирует практически изотермически. Доля общей энтальпии системы, которая способна производить работу при изотермических условиях, обозначается как свободная энтальпия(G) (англ. Gibbs' free energy — свободная энергия Гиббса). Приведем основное уравнение отношения между изменениями энтропии и энтальпии и изменением свободной энтальпии:
∆G = ∆Н - Т ∆S. (6.3)
1 В некоторых случаях колебания температуры значительны, как, например, при термогенезе в соцветиях ароидных. Повышение температуры связано с экзергоническими реакциями при дыхании (см. далее). — Примеч. ред.
При этом ∆G — изменение свободной энтальпии системы; изменение энтальпии ∆Н — теплота, которая обменивается между системой и окружающей средой в том случае, когда система не совершает работу (см. выше); Т — абсолютная температура (К); ∆S — изменение энтропии системы.
Знак ∆G позволяет решить, может ли данная реакция протекать самопроизвольно или нет. Если ∆G > 0, реакция не протекает самопроизвольно, и осуществляется только при ∆G < 0 (хотя не обязательно быстро). Свободно протекающая реакция происходит с уменьшением свободной энтальпии и увеличением энтропии так долго, пока АН не уравняется с Т ∆S и таким образом будет достигнуто состояние ∆G = 0 (состояние равновесия). Процессы, в которых ∆G < 0, называются экзергоническими, а при ∆G > 0 — эндергоническими.
При Т = 0 или ∆S = 0 ∆G реакции может быть определена из теплового эффекта реакции (т.е. изменения энтальпии ∆Н). Эти условия, однако, не выполняются в биологических системах. Тем не менее ∆G и с ней движущую силу реакции можно приближенно определить из теплового эффекта реакции, когда значение ∆Н велико, как, например, при окислении питательных веществ (см. 6.10.3) в дыхании, а температура невысока (как это часто происходит в клетках), так что компонент T∆S мало влияет на значение свободной энтальпии. Однако в процессах с низким тепловым эффектом, таких, как гидролитическое расщепление, а также при полимеризации (конденсации) — т.е. столь же важных биологических реакциях — изменение энтропии может существенно определять свободную энтальпию. Так как ∆S выступает в комплексе с Температурой, влияние энтропии на ∆G возрастает пропорционально абсолютной температуре.
Для понимания течения химических реакций целесообразно связать свободную энтальпию реакции с изменением количества веществ и в конечном счете — с устанавливающимся в реакции равновесием. Возможность этого подтверждается тем, что химическая реакция А → В, при уменьшении свободной энергии будет протекать так долго, пока минимум энтальпии (∆G = 0) не будет достигнут. Вслед за этим дальнейшего абсолютного изменения количества вещества уже не происходит, устанавливается равновесие А ⇄ В и соотношение концентраций конечного продукта и исходного вещества остается постоянным (закон действующих масс). Это соотношение называется термодинамической константой равновесия, К:
![]() (6.4)
(6.4)
а в общем случае для реакции А + В ⇄ С + D:
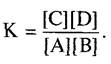 (6.5)
(6.5)
Константа равновесия, таким образом, задается через произведение концентраций продуктов реакции, деленных на произведение концентраций исходных веществ в состоянии равновесия. Отношение между ∆G и К выражается уравнением
∆G0 = RТ In К (Дж • моль-1), (6.6)
причем ∆G0 есть изменение стандартной молярной свободной энтальпии (на моль превращенного вещества при стандартных условиях: температуре Т = 25 °С и р = 1 атм = = 0,1 МПа), Т — абсолютная температура в Кельвинах (К), R — универсальная газовая постоянная (= 8,314 Дж • моль-1 - К-1).
Для реакций, в которых принимают участие ионы водорода, — как это часто имеет место в биологических системах, их стандартную концентрацию также нужно принять равной 1 моль. В биохимической литературе из практических соображений при определении стандартных условий принято указывать не количество вещества в молях, а изменение молярной концентрации (моль л-1). Это означает, что для стандартного состояния изменение концентрации ионов водорода окажется равным 1 моль • л-1 (pH 0), что является совершенно не физиологической величиной. Поэтому в биохимической литературе употребляется несколько иное определение стандартного состояния, в котором концентрация Н+-ионов равна 10-7М (pH 7), а концентрация воды (55,5 моль • л-1), практически не изменяющаяся при протекании реакции, вводится в число констант (если вода присутствует в уравнении реакции):
∆G0' = RТ In К'. (6.7)
Таблица 6.3. Изменения молярной свободной стандартной энтальпни при pH 7 (∆G0') для некоторых важных реакций обмена веществ (реакций гидролиза)
Реакция |
∆G0, кДж • моль-1 |
Фосфоенолпируват + Н2O —» пируват + Фн1 |
-61,9 |
1,3-Дифосфоглицерат + Н2O —» 3-фосфоглицерат + Фн |
-49,4 |
Пирофосфат + Н2O —» 2ФН |
-33,5 |
АТФ + Н2O —» АМФ + ФФН |
-32,2 |
АТФ+Н2O—» АДФ + Фн |
-30,5 |
Глюкозо-1 -фосфат + Н2O —» глюкоза + Фн |
-20,9 |
Глюкозо-6-фосфат + Н2O —» глюкоза + ФH |
-13,8 |
Глицерин-З-фосфат + Н2O —» глицерин + Фн |
-9,2 |
1 В отечественной литературе допускается вместо Фн обозначать неорганический фосфат Pi (от англ. inorganic). — Примеч. ред.
Изменения стандартной молярной свободной энтальпии (при pH 7) некоторых важных реакций приводятся в табл. 6.3.
В клетке, однако, господствуют условия, которые отличаются от стандартных. Так, pH часто отклоняется от 7,0, температура — от 25 °С, и в особенности концентрации веществ, как правило, не соответствуют стандартным условиям. Следует поэтому тщательно различать изменение стандартной молярной свободной энтальпии ∆G0', которая постоянна при данной температуре, и действительное изменение свободной энтальпии ∆G, которое зависит от реальной температуры и реальных концентраций компонентов реакции. Не ∆G0', а ∆Gопределяет направление протекания реакции в клетке. Во многих случаях, однако, установить эти значения ∆G чрезвычайно сложно, так как реальные условия (концентрации веществ, значения pH, температура) в отдельных реакционных пространствах (компартментах) трудно определить точно.
6.1.3. Энергетика открытых систем
Из термодинамики закрытых или равновесных систем следуют важные заключения относительно энергетики отдельных биохимических реакций (особенно полезным может быть заключение о том, способен ли вообще определенный процесс протекать самопроизвольно или же нет), однако живые организмы — это открытые системы, которые постоянно обмениваются энергией и веществами с окружающей средой (см. рис. 6.1). В то время как любая закрытая система стремится к стационарному равновесному состоянию (∆G = 0), открытые системы способны сохранять стабильное состояние, весьма далекое от термодинамического равновесия, т.е. стационарное состояние (англ. steady state). Термодинамическое описание подобных открытых систем относится к задачам неравновесной, или необратимой, термодинамики, в которой прежде всего учитывается временной фактор и большую роль играют потоки веществ. Здесь нет возможности детально останавливаться на необратимой термодинамике, но понятие химического потенциала (см. 6.1.4) оказывается весьма полезным, чтобы лучше понять энергетику многих физиологических процессов.
Для стабильного стационарного состояния характерно, что поток веществ и энергии через систему постоянно создает в ней свободную энтальпию. Это происходит в конечном счете посредством экзергонического превращения органических соединений (питательных веществ) с высокой энтальпией и более низкой энтропией в «продукты распада» с низкой энтальпией и более высокой энтропией (см. 6.10). Фотосинтетически активные клетки производят эти продукты (первичную продукцию) прежде всего из неорганических соединений при помощи поглощенной энергии света в высокоэкзергоническом процессе фотосинтеза (см. 6.4—6.7). Свободная энтальпия в форме богатых энергией соединений, как, например, АТФ, употребляется для совершения биологической работы и поддержания высокого уровня упорядоченности, характерного для живых существ. Если поток веществ и энергии прерывается, то через некоторое время устанавливается стационарное состояние равновесия (∆G = 0) — наступает смерть.
Было показано, что стационарное состояние — это состояние открытой системы, в котором она создает минимальную энтропию и в котором может поддерживаться максимально возможная упорядоченность при минимальных затратах энергии. Стационарное состояние, таким образом, — это состояние открытой системы с максимальной термодинамической эффективностью. Существенно, что в противоположность системе в стационарном равновесии, каким оно устанавливается для закрытой системы, система в стационарном состоянии может регулироваться (важное свойство всех живых клеток).
6.1.4. Химический потенциал
Свободную энтальпию открытой системы со сложным составом, которую представляет собой клетка, практически нельзя определить. Однако во многих случаях достаточно установить способность определенных компонентов этой системы производить работу. Так, например, представляет интерес лишь разность свободной энтальпии протонов (но не других ионов) с двух сторон клеточной мембраны, если удается рассчитать движущую силу протонов для выполнения работы в сопряженных транспортных процессах и их направление. Или же интересно выявить различие свободной энтальпии воды в соседних водных растворах, разделенных клеточной мембраной, направление и масштабы потока воды через эту поверхность раздела.
Свободная энтальпия, рассчитанная на моль компонента I в смеси веществ из к компонентов, называется химическим потенциалом μ компонента i (μ). Сумма химических потенциалов всех к компонентов дает свободную энтальпию на моль смеси веществ. Таким образом, вклады отдельных компонентов взаимно дополняются. Химический потенциал каждого компонента смеси веществ, в свою очередь, можно разложить на стандартный потенциал (μ0) и сумму составляющих, отражающих отклонения от стандартного состояния:
![]()
где RT In х1 — концентрационная составляющая: R — универсальная газовая постоянная; Т — абсолютная температура; х1 — мольная доля i (х1 = n1: (nа + nb ... nк)). Мольная доля — это отношение количества вещества (в молях) данного компонента к общему количеству всех веществ, находящихся в растворе, включая растворитель; рV1 — составляющая давления: р — давление, V1 — парциальный молярный объем i, соответствует изменению объема системы при добавлении 1 моля компонента i; ghM1 — гравитационная составляющая: g — гравитационная постоянная (9,806 м c-2), h — высота подъема, М1 — молярная масса i; FЕz1 — электрическая составляющая: F — постоянная Фарадея (96,49 кДж • В-1 • моль-1), Е — электрический потенциал, z1 — величина заряда i.
Размерность μ выражается в единицах энергии на моль (Дж • моль-1).
Поскольку часто представляет интерес не химический потенциал как таковой, а его изменение при изменении состояния системы относительно компонента i, то для расчета изменения химического потенциала (= свободной энтальпии) i в смеси при переходе состояний А → В выводится соотношение
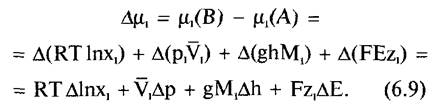
Варианты общих уравнений 6.8 и 6.9 представляют интерес для следующих глав и будут рассмотрены ниже.
Поскольку растительные клетки, как и клетки других организмов, не могут активно транспортировать воду, она перемещается пассивно из участка с более высокой (более положительной) свободной энтальпии к участку с более низкой (более отрицательной) свободной энтальпией, т.е. это экзергонический процесс, протекающий самопроизвольно (но не обязательно быстро). Так как в биологическом аспекте существенны лишь смеси воды с другими веществами (например, в клетках и почве — водные растворы, в воздушной среде — водяной пар в газовой смеси), то в энергетических расчетах целесообразно применять понятие водного потенциала, или потенциала воды (μн2о)- Молекулы воды не заряжены электрически (ZH2O = 0), поэтому из уравнения 6.9 выпадает электрическая составляющая и оно принимает вид
![]()
Следовательно, для чистой воды (хH2O = 0) в стандартном состоянии (р = 0, h = 0) значение μH2O = μ0H2O.
Согласно соотношению хН2о=Σ1х1=1, можно выразить концентрационную составляющую RТ In хН2о как функцию мольной доли всех растворенных частиц RТ In (1 - Σ1х1). Для разбавленных растворов можно приближенно подставить 1n (1 - х) = -х; применение соотношения Σ1Х1 =VH2OΣ1C1 где с — молярная концентрация, дает в итоге уравнение
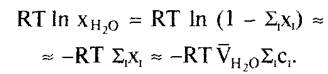
Для более концентрированных растворов (как правило, 0,1 М и выше) следует употреблять моляльные концентрации (моль кг1) и активности вместо концентраций.
Теперь RТ Σ1C1= П (П — осмотическое давление, закон Ван-Гоффа), что дает
![]()
и отсюда
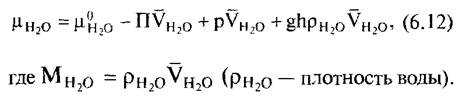
Поскольку и в этом случае разность химических потенциалов воды, как правило, представляет больший интерес, чем абсолютная величина потенциала, то при дополнительном преобразовании относительно парциального молярного объема воды в первую очередь определяют отклонение химического потенциала воды в рассматриваемой системе от стандартного состояния
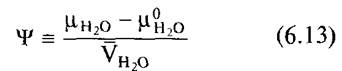
как водный потенциал раствора. Далее из уравнения 6.12 следует
![]()
имеет размерность энергия/объем (=сила/площадь = давление) и выражается в единицах бар или Па (1 бар = 0,1 МПа).
В клеточных масштабах различие в высоте не значительно, так что уравнение 6.14 после сокращения гравитационной составляющей упрощается далее до
![]()
Водный потенциал раствора, т.е. свободная энтальпия воды на парциальный молярный объем воды (VН2O - 18 мл), определяется, таким образом, тремя составляющими потенциалами:
✵ потенциал давления, р (гидростатическое давление, при котором раствор неподвижен);
✵ осмотический потенциал, -П (отрицательное значение осмотического давления П);
✵ гравитационный потенциал (последним при рассмотрении процессов в клеточных масштабах можно пренебречь).
Следует отметить, что гидростатическое давление определяется как отклонение от давления окружающей среды. Оно может принимать как положительные значения («избыточное давление»), так и отрицательные («разрежение», «откачивание»). Абсолютное давление всегда положительно и в полном вакууме соответственно равно нулю. Следовательно, потенциал давления воды (р) в стандартном состоянии равен 0 (р = 0), а ее абсолютное давление составляет 1 бар (0,1 МПа).
Если между водными потенциалами двух компартментов имеется различие ∆![]() '=0), то вода будет постоянно двигаться из зоны с более положительным водным потенциалом в зону с более отрицательным водным потенциалом. При этом снижается ее свободная энтальпия. Это также экзергонический процесс и, следовательно, он протекает самопроизвольно.
'=0), то вода будет постоянно двигаться из зоны с более положительным водным потенциалом в зону с более отрицательным водным потенциалом. При этом снижается ее свободная энтальпия. Это также экзергонический процесс и, следовательно, он протекает самопроизвольно.
Понятие водного потенциала и следствия этой концепции оказываются весьма полезными для понимания всего водного обмена растения (см. 6.3).
6.1.4.3. Химический потенциал ионов и трансмембранный потенциал
Химический потенциал электрически заряженных частиц в растворе определяется главным образом их концентрацией и электрическим зарядом. Соответственно этому записывается уравнение химического потенциала для иона i:
![]()
(а1 - активность иона I; для разбавленных растворов а1∞ с1; с1 — молярная концентрация i).
Если рассматривать два раствора i в компартментах А и В, разделенных электроизолирующей мембраной, то разность химических потенциалов i, ∆μ1 (называемая также электрохимическим потенциалом), определяется как![]()
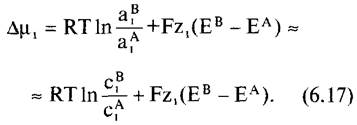
Особенное значение в дальнейшем будет иметь электрохимический потенциал ионов водорода на мембранах клетки, так как он представляет собой движущую силу многих транспортных процессов через клеточные мембраны и для синтеза АТФ в хлоропластах (см. 6.4.9), а также и митохондриях (см. 6.10.3.3). Для Н+ ZH+= 1, и отсюда имеем
![]()
При этом реакционное пространство А представляет внутриклеточный, а реакционное пространство В — внеклеточный (или функционально внеклеточный) компартменты. Разность потенциалов Ев - ЕА = ∆ЕМ обозначается как трансмембранный электрический потенциал (кратко: мембранный потенциал). В упрощенной форме можно записать и при сведении вместе всех констант при стандартной температуре (Т = 298 К), применяя определение значения pH (pH = -log[Н+]), получаем
![]()
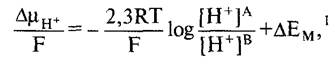
1 Здесь и далее log — десятичный логарифм. В отечественной литературе его обычно обозначают символом lg. — Примеч. ред.
Выражение![]() обозначается как протон-движущая сила (англ. proton motive force — pmf) и используется для характеристики энергии протонного градиента. При этом две составляющие, каждая по отдельности или совместно, способны выполнять работу: с одной стороны, концентрационный потенциал ионов водорода (∆рН), с другой — электрический потенциал (∆ЕМ). Примеры см. в 6.1.5.
обозначается как протон-движущая сила (англ. proton motive force — pmf) и используется для характеристики энергии протонного градиента. При этом две составляющие, каждая по отдельности или совместно, способны выполнять работу: с одной стороны, концентрационный потенциал ионов водорода (∆рН), с другой — электрический потенциал (∆ЕМ). Примеры см. в 6.1.5.
Для состояния равновесия (∆μ1 = 0) из уравнения 6.17 получаем
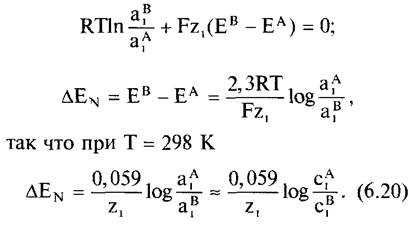
Это уравнение называется уравнением Нернста (∆ЕN, равновесный потенциал Нернста, В).
Для![]() между обоими компартментами возникает разность потенциалов. При эффективной 10-кратной разности концентраций (при z1 =1) разница напряжений составит 59 мВ (при z1 = 2 соответственно 29,5 мВ). Верно обратное: при приложенном постоянном напряжении в 59 мВ. в состоянии равновесия для проницаемого иона между этими компартментами будет устанавливаться разница концентраций в 1:10.
между обоими компартментами возникает разность потенциалов. При эффективной 10-кратной разности концентраций (при z1 =1) разница напряжений составит 59 мВ (при z1 = 2 соответственно 29,5 мВ). Верно обратное: при приложенном постоянном напряжении в 59 мВ. в состоянии равновесия для проницаемого иона между этими компартментами будет устанавливаться разница концентраций в 1:10.
6.1.4.4. Окислительно-восстановительный потенциал
Многочисленные имеющие отношение к биологии превращения веществ протекают с восстановлением или окислением метаболитов. Под восстановлением понимают присоединение электронов, под окислением — отдачу электронов молекулой. Окисление и восстановление протекают, как правило, как сопряженные процессы (окислительно-восстановительные, или редоксреакции). Окислительно-восстановительные реакции можно описать также с помощью определенного для ионов в уравнении 6.17 химического потенциала (электрохимического потенциала). При этом для сопряженных реакций Аox + Вгеd ⇄ Агеd + Вох уравнение Нернста принимает следующий вид:
![]()
где R, Т и F — введенные ранее величины; z — число перенесенных согласно формуле реакции электронов; ∆Е0 — разница стандартных окислительно-восстановительных потенциалов окислителя и восстановителя: ∆Е0 = Еов - Е0А. Они определяются для восстановителя и окислителя как разница потенциалов относительно нормального водородного электрода при стандартных условиях (потенциал которого принимают равным 0) и сами представляют, таким образом, разность потенциалов.
Е0- или ∆Е0-значения, как обычно, стандартизуются к 25 °С, давлению в 1 атм (0,1 МПа) и для 1 моль • л-1 изменению вещества. Если в окислительно-восстановительных реакциях принимают участие ионы водорода (протоны), то соответственно имеет место изменение количества вещества в 1 моль/л (pH 0, см. уравнение 6.6). Для биологических целей в силу этих обстоятельств выбирается также и здесь, как ранее для ∆G0, иное определение стандартных условий (Е0): pH 7. Существует зависимость
Е0' = Е0 - 0,42 В. (6.22)
Некоторые стандартные потенциалы для pH 7 сведены вместе в табл. 6.18 (см. 6.4.5).
Редокс-потенциал ∆Е выражает электрохимическую энергию, которую окислительно-восстановительная реакция поставляет для выполнения работы на один перенесенный моль электронов. Разница свободной энтальпии реакции может быть легко определена из редокс-потенциала через отношение:
∆G - -zF • ∆E. (6.23)
Соответственно
∆G0' = -zF • ∆Е0'. (6.24)
Из стандартных редокс-потенциалов может быть определено направление, в котором для сопряженных окислительно-восстановительных реакций процесс будет протекать самопроизвольно (но не обязательно быстро) при стандартных условиях. Окислительно-восстановительная реакция экзергонична (∆G < 0), если электроны переходят от партнера реакции с более отрицательным стандартным редокс-потенциалом (т.е. восстановителем, который окисляется в процессе реакции) к участнику реакции с более положительным стандартным редокс-потенциалом (т. е. окислителю, который восстанавливается в течение реакции). Так как в клетке, однако, господствуют не стандартные условия, анализ значений ∆Е0 не обязательно отражает действительный ход реакции в клетке. Для этого необходимо знание ∆Е (и отсюда ∆G), что предусматривает прежде всего знание действительных концентраций, участвующих в окислительно-восстановительной реакции (редокс-реакции) компонентов, а также реальной температуры и значения pH. Эти факторы, однако, как правило, не известны абсолютно точно, так что для рассмотрения принципиальных энергетических отношений окислительно-восстановительных реакций, как и вообще в биохимических процессах, часто оперируют стандартными значениями.
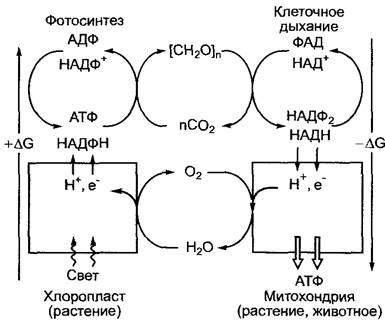
Окислительно-восстановительные реакции играют в обмене веществ центральную роль. Как фотосинтез, так и клеточное дыхание представляют собой окислительно-восстановительные процессы (рис. 6.2). При фотосинтезе углерод восстанавливается с уровня окисления СO2 (степень окисления +4) до уровня восстановления углеводов ([СН2O]n, степень окисления 0). Электроны поставляются из воды и через сложную, движимую светом (эндергоническую) окислительно-восстановительную цепь переносятся вначале на окисленный никотинамидадениндинуклеотидфосфат (НАДФ+) с образованием НАДФН, который служит транспортной молекулой для восстановительных эквивалентов и вновь окисляется в реакциях ассимиляции СO2 (см. 6.5.2). Дыхательные процессы в митохондриях также включают окисление углеводов до уровня СO2, чтобы использовать полученные электроны для второй широко распространенной молекулы, транспортирующей окислительно-восстановительные эквиваленты, восстановленного никотинамидадениндинуклеотида (НАДН) из окисленного никотинаденин- динуклеотида (НАД+), и последующий транспорт электронов через мембранные комплексы к кислороду (см. 6.10.3.3.) Помимо этих двух фундаментальных окислительно-восстановительных процессов в обмене веществ играют роль также другие многочисленные окисления и восстановления метаболитов, которые катализируются окислительно-восстановительными ферментами (оксидоредуктазами).
Рис. 6.2. Энергетические принципы двух фундаментальных метаболических процессов биосферы, фотосинтеза и клеточного дыхания.
Серым выделены окислительно-восстановительные процессы, протекающие на мембранных системах, которые служат превращению энергии (фотосинтез, см. 6.4; клеточное дыхание, см. 6.10.3)
6.1.5. Превращение энергии и энергетическое сопряжение
Из законов термодинамики следует, что изменение свободной энтальпии (∆G) какой-либо серии сопряженных процессов (к примеру, химических реакций) равно сумме изменений свободной энтальпии отдельных реакций. Это имеет важные следствия для обмена веществ, так как многочисленные эндергонические процессы метаболизма могут протекать самопроизвольно лишь тогда, когда за счет сопряжения с экзергоническими реакциями изменение свободной энтальпии процесса в целом отрицательно (∆G < 0), вся реакция, таким образом, протекает экзергонически. Это обозначают как энергетическое сопряжение. Оно выступает в первую очередь в последовательных биохимических цепях и характерно практически для всего обмена веществ. Специфические доставляющие энергию реакции многократно используются в метаболизме, чтобы привести в действие сильно эндергонические реакции. Б клетках энергия запасается чаще всего в виде аденозинтрифосфата (рис. 6.3; структуру см. на рис. 1.3).
Рис. 6.3. Энергетическое сопряжение экзергонических и эндергонических реакций в метаболических процессах при участии аденилат- ной системы (АТФ, АДФ + Фн) на примере сопряжения гидролиза фосфоенолпирувата с фосфорилированием глюкозы до глюкозо-6- фосфата
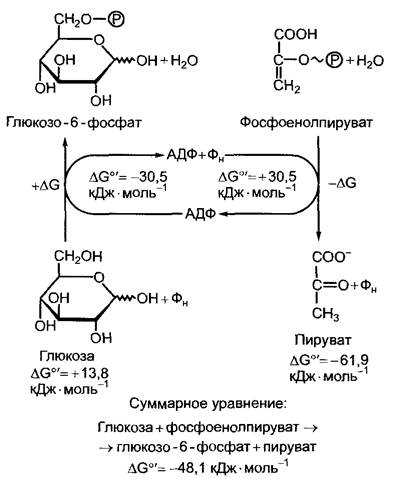
До определенной степени при специфических биосинтезах (например, нуклеиновых кислот см. 1.2, углеводов см. 6.17.1, липидов см. 6.11) могут использоваться и другие богатые энергией нуклеозидтрифос- фаты. Реакция гидролиза АТФ + Н2O -» АДФ + Фн (Фн — неорганический фосфат) является сильно экзергоническим процессом, что можно видеть по значению стандартной молярной свободной энтальпии (при pH 7) ∆G0' = -30,5 кДж-моль-1. Образование АТФ по схеме: АДФ + Фн
АТФ + Н2O в силу этих причин сильно эндергонично: ∆G0' = +30,5 кДж-моль-1. АТФ может образоваться за счет сопряжения с подходящей экзергонической реакцией. При этом донор фосфатной группы должен обладать по меньшей мере равной энергией фосфатной группы, т.е. при гидролизе должен обеспечивать столь же высокое значение свободной энтальпии. Это происходит при гидролизе, например, 1,3- дифосфоглицерата или фосфоенолпирувата (ФЕП) (см. табл. 6.3; рис. 6.3). Для таких реакций образования АТФ употребляется понятие «фосфорилирование на уровне субстрата», или «субстратное фосфорилирование». Преобладающая доля АТФ растительной клетки, однако, синтезируется хемиосмотически,т.е. за счет энергетического сопряжения с протонным градиентом, который образуется в митохондриях при окислении молекул субстрата (см. 6.10.3.3) и в хлоропластах в процессе световых реакций фотосинтеза (см. 6.4.9).1
1 В этом случае используют термин «мембранное фосфорилирование», «фосфорилирование на уровне мембран». — Примеч. ред.
Таким образом, вторая возможность энергетического сопряжения эндергонических реакций с экзергоническими состоит в использовании электрохимической энергии ионных градиентов (рис. 6.4). У растений это градиенты ионов водорода (протонов), которые существуют на плазмалемме, тонопласте и мембранных системах митохондрий и хлоропластов. В двух последних случаях они служат, как было упомянуто, для синтеза АТФ; протонные градиенты на плазмалемме и тонопласте продуцируются за счет гидролиза АТФ протон-транспортирующими АТФазами (Н+-АТФазами, протонными помпами). В дополнение к этому на тонопласте существует протонная помпа, которая использует энергию гидролиза пирофосфата. Транспортные процессы, как в данном случае транслокация ионов водорода через клеточную мембрану, которые механически сопряжены с гидролизом богатой энергией связи (в данном случае — фосфоангидридной связи), называются первично активными транспортными процессами. Протон-движущая сила при использовании градиента состоит, как упомянуто (см. уравнение 6.19), из двух частных потенциалов: электрического и концентрационного. Этот электрохимический потенциал ионов водорода поставляет движущую силу для сопряженного с ним (эндергонического) транспорта других ионов или электронейтральных метаболитов (вторично активный транспорт). При этом может использоваться или электрическая составляющая протон-движущей силы (например, при ионном транспорте через зависящие от напряжения ионные каналы — электрическое сопряжение), или же как электрический, так и концентрационный протонный градиент (электрохимическое сопряжение), как, к примеру, при транспорте ионов водорода с электрически нейтральными метаболитами через переносчики-транспортеры (транслокаторы — англ. carrier). Ионные каналы, как правило, проводят в обоих направлениях и селективны для отдельных ионов или по крайней мере родственных типов ионов; переносчики главным образом высокоселективны для своего субстрата. По направлению транспорта различают унинортеры, симпортеры и антипортеры (см. рис. 6.4). Наиболее важные из известных сейчас первично или вторично активных транспортных систем плазмалеммы и тонопласта сведены вместе на рис. 6.5.
Рис. 6.4. Энергетическое сопряжение экзергонических и эндергонических реакций через ионные градиенты на клеточных мембранах (на примере плазмалеммы).
В отношении растений речь идет о градиентах ионов водорода. Эндергоническое накопление протонов во внеклеточном пространстве идет за счет сильно экзергонического гидролиза АТФ. Произведенная при этом протон-движущая сила используется для вторично активных процессов ионными каналами (двойные блоки) или транспортерами (переносчики, транслокаторы, carrier) (круги). Их подразделяют на унипортеры, симпортеры и антипортеры в зависимости от того, транспортируется ли только одна частица (унипорт) или же две частицы — в одном направлении (симпорт) либо в противоположных направлениях (антипорт). Ионные каналы и транспортеры в принципе могут быть задействованы также в пассивном транспорте, при котором должен присутствовать лишь концентрационный градиент транспортируемых частицы или частиц. Такие транспортные процессы, протекающие лишь до выравнивания концентраций и опосредованные белками, называют облегченной диффузией. Пример пассивного переносчика — триозофосфатфосфатный транслокатор внутренней мембраны хлоропласта (см. рис. 6.73). Пассивными каналами являются также порины внешних мембран хлоропластов и митохондрий, а также мембраны пероксисом (или глиоксисом) (см. 6.5.6; 6.12).
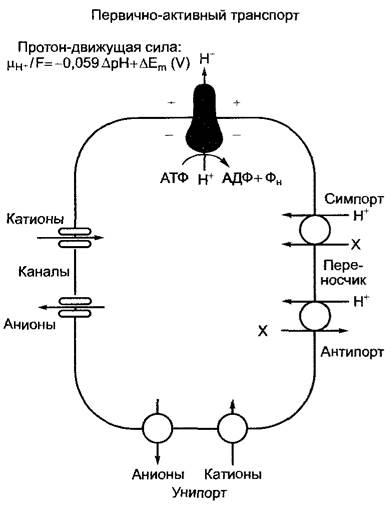
Рис. 6.5. Примеры разнообразных первично активных и пассивных, или вторично активных (показано черным), транспортных процессов на плазмалемме и тонопласте растительных клеток. Стехиометрия симпортеров и антипортеров не во всех случаях известна, рисунок передает лишь тип транспортируемой частицы и направление переноса. АТФазы P-типа образуют фосфорили- рованный интермедиат в процессе транспортного цикла (Р-фосфоинтермедиат); АТФазы V-типа напоминают по своему строению АТФ-синтазы митохондрий (F1/F0-АТФаза) или хлоропластов (CF1/CF0-АТФаза) (V — от вакуолярная). ABC-транспортеры используют энергию АТФ для транслокации более крупных органических соединений и комплексов, таких, как фитохелатин-Сd2+-комп- лекс (PC-Cd2+) или конъюгаты антоцианов с глутатионом (антоциан-GS). ABC-транспортеры характеризуются присутствием специальной аминокислотной последовательности, которая требуется для связывания АТФ (АВС = ATP-binding cassette)
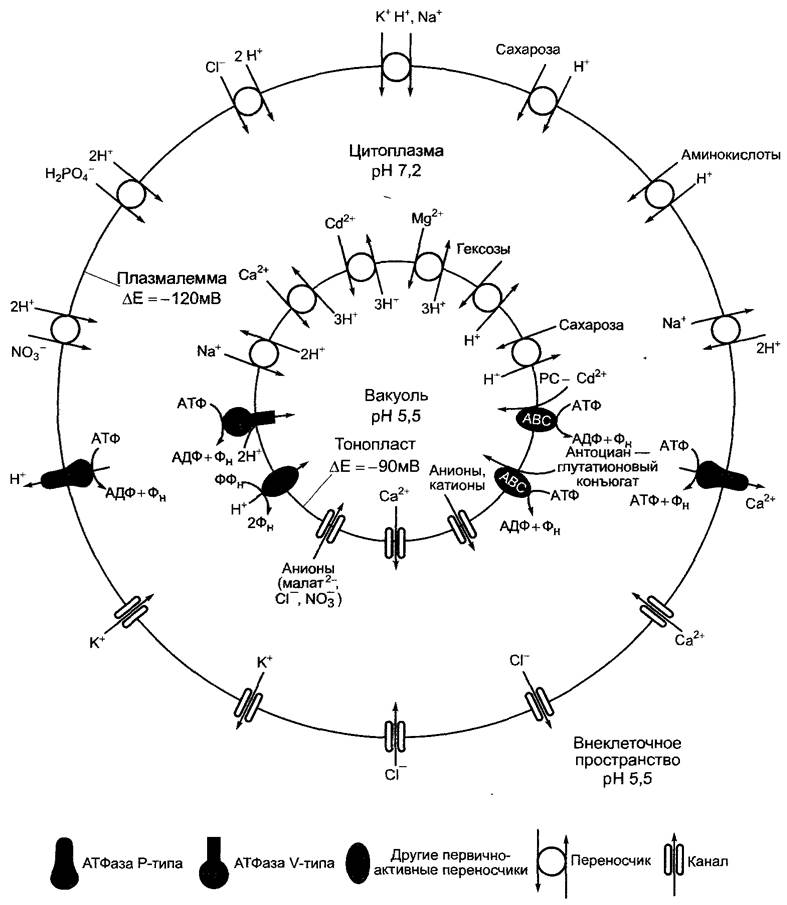
Рис. 6.6. Сопряжение механических и химических процессов в моторных белках на примере смещения микротрубочек, осуществляемого динеином (подвижность жгутиков и ресничек, см. 8.2).
Динеин — очень большой белковый комплекс с молекулярной массой 1 — 2 • 106 Да, который содержит от двух до трех силообразующих головок (представлена только одна), являющихся частью тяжелой субъединицы динеина и несущих АТФазную активность. Для наглядности представлена только тяжелая субъединица динеина, состоящая из основания, головки и ручки; головка и ручка образуют вместе моторный домен динеина. В случае аксонемной структуры жгутиков динеин связывается с А-трубочками, палочкообразные ручки динеиновых головок в отсутствие АТФ прочно связываются со специфическими сайтами на В-трубочках. Связывание и гидролиз АТФ в головке динеина приводят к изменению конформации, которая передается на ручки. В результате ручка на короткое время освобождается от В-трубочки, передвигается в направлении минус-конца В-трубочки (к основанию жгутика) и образует новый контакт с молекулой тубулина. При диссоциации АДФ и Фн восстанавливается исходная конформация динеина и связанное состояние ручки на В-трубочке. На этом этапе происходит передача механического усилия с динеина на В-трубочку, которая сдвигается относительно А-трубочки в направлении плюс-конца. Так как в аксонеме трубочки связаны нексиновыми мостиками и закреплены в основании, скользящее движение микротрубочек приводит к искривлению жгутика
Исходным предназначением протонных помп плазмалеммы и тонопласта предположительно было установление цитоплазматического значения pH растительной клетки в узкой области значений между 7,5 и 8,0, в неравновесии в основном к кислым внешней среде и содержимому вакуоли. У морских водорослей хлорид накачивается внутрь клетки за счет действия СI--транспортирующей АТФазы (хлоридной помпы); в соляных железках (Limonium, Tamarix), напротив, СI- выводится; Na+ следует за ним по механизму электрического сопряжения.
Са2+-транспортирующие АТФазы плазмалеммы и эндоплазматического ретикулума отвечают за выведение пассивно диффундирующего в клетку Са2+ из цитоплазмы и таким образом поддерживают низкий уровень Са2+ в цитоплазме (порядка 1(Н М).
Третья возможность энергетического сопряжения состоит в сохранении свободной энтальпии в форме активированной конформации молекулы белка, переход которой в более бедное энергией основное состояние используется для выполнения работы. Моторные белки, как, к примеру, динеин, превращают энергию фосфоангидридной связи АТФ в механическую энергию; при этом в качестве активированной конформации выступает фосфорилированная форма белка (рис. 6.6). Транс-локация ионов АТФазами основана на различной конформации нефосфорилированных и фосфорилированных молекул фермента. За счет этого в реакционном цикле на обеих сторонах мембраны экспонируются ион-связывающие сайты с различной аффинностью.
Энергетическое сопряжение, как уже стало ясно на основании нескольких примеров, часто протекает при конверсии энергии. Так, растения переводят энергию солнечного света вначале в электрохимическую (электрическое разделение зарядов и концентрационный потенциал ионов водорода), а в заключение — в химическую форму (НАДФН, АТФ — см. 6.4.4, рис. 6.2). Моторные белки конвертируют химическую энергию в механическую, транспортирующие ионы АТФазы переводят химическую энергию в электрохимический потенциал, и электрохимический потенциал ионных градиентов (в растениях — градиентов ионов водорода) за счет разнообразнейших процессов транспорта веществ используется для выполнения осмотической работы (концентрирования веществ против падения электрохимического потенциала).
6.1.6.1. Основные принципы катализа
Равновесная термодинамика позволяет предсказать, возможна ли энергетически определенная реакция при данных условиях, т. е. может ли она протекать самопроизвольно. Она позволяет также рассчитать концентрации участников реакции, наблюдаемые при равновесии. Однако равновесная термодинамика не дает указаний о скорости, с которой протекают самопроизвольные реакции. В действительности же эта скорость может быть крайне мала. Так, к примеру, окисление глюкозы кислородом — весьма экзергонический процесс, однако при физиологических температурах и нормальном давлении глюкоза остается в присутствии кислорода практически неограниченно стабильной. Основная причина заключается в том, что участники химических реакций должны находиться в активирован
ном состоянии, из которого и происходит реакция. Перевод реагирующих веществ из основного в это богатое энергией состояние требует притока энергии. Количество энергии, требующееся на моль вещества, называется молярной свободной энтальпией активации(∆G+), часто также кратко называемой энергией активации. При химических реакциях активация может произойти за счет повышения температуры. Оно увеличивает число реакционноспособных молекул и тем самым ускоряет реакцию (в основном в два раза при повышении температуры на 10 °С). Биохимические реакции должны, однако, протекать при сравнительно низких температурах. К тому же в живых существах происходят только очень незначительные изменения температуры, метаболические процессы протекают практически изотермно. Повышение температуры как средство ускорения реакций обмена веществ поэтому исключается.
Катализаторы (греч. kata — вниз, lysis — разложение) — это вещества, добавление которых к реакционной смеси повышает скорость реакции, причем энергия активации оказывается пониженной (рис. 6.7). Катализаторы выходят из реакции неизмененными и не влияют на положение реакционного равновесия (таким образом, не изменяют свободной энтальпии реакции, ∆G). Они всего лишь ускоряют реакцию и, таким образом, достижение состояния равновесия. Так как при реакции они не изменяются и не используются, то могут использоваться вновь и вновь для выполнения своей задачи и должны быть в наличии лишь в малых количествах.
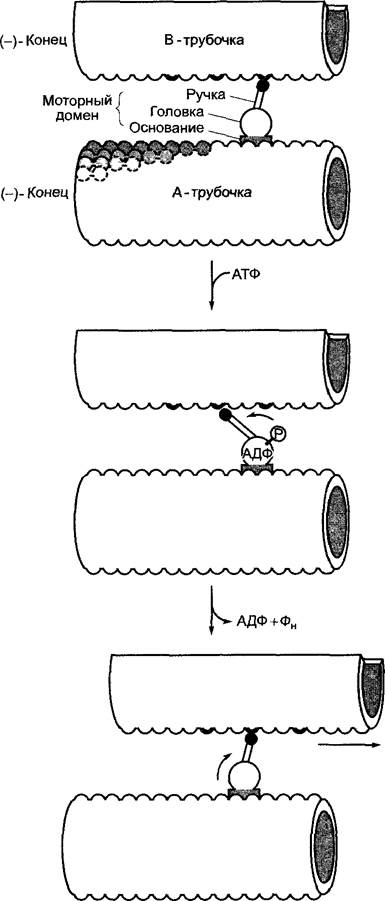
Рис. 6.7. График изменения во времени энергии системы для некатализируемой и катализируемой реакций на примере диспропорционирования Н2O2.
Для катализируемой ферментом реакции (показано серой линией) протекание реакции показано более точно. Длина стрелок на графике пропорциональна соответствующей свободной энтальпии реакции диспропорционирования Н2O2; Е — фермент; S — субстрат; ЕS — фермент-субстратный комплекс; ЕР — комплекс фермент-продукт; Р — продукт реакции
Ускорение реакций метаболизма является задачей биокатализаторов. За исключением нескольких каталитически активных рибонуклеиновых кислот (рибозимов), биокатализаторы — это белки. Они обозначаются как энзимы (греч. zymе — дрожжевое тесто) или ферменты (лат. fermentum — дрожжевое тесто) и подчиняются тем же самым закономерностям, что и химические катализаторы; энзимы также понижают энергию активации катализируемой реакции (рис. 6.7), не изменяя равновесия реакции (и при этом ∆G).
Так, молярная свободная энтальпия активации (∆G+ реакции диспропорционирования перекиси водорода Н2O2 -» Н2O + 1/2O2 : ∆G++ = +75 кДж • моль-1; это значение может быть достигнуто за счет нагревания раствора Н2O2. В присутствии мелкоизмельченной платины ∆G++ = +49 кДж • моль-1. Платина выступает в роли катализатора, и реакция протекает с измеримой скоростью уже при комнатной температуре. Фермент каталаза осуществляет диспропорционирование с энергией активации ∆G+ = +23 кДж-моль-1. В присутствии каталазы перекись водорода при комнатной температуре стремительно разлагается на Н2O + 1/2O2. В каждом случае молярная стандартная энтальпия экзергонической реакции составляет ∆G+ = -97 кДж • моль-1. Ферменты — чрезвычайно эффективные катализаторы. Так, карбоангидраза ускоряет гидратацию СO2, согласно уравнению Н2O + СO2 ⇄ Н2СO3, в 107 раз при обороте 105 молекул СO2 в секунду на одну молекулу фермента.
Ферментативная реакция (см. рис. 6.7) протекает вначале с образованием комплекса фермент-субстрат (ЕS); этот комплекс превращается в комплекс фермента с продуктом (ЕР), который быстро диссоциирует с высвобождением продуктов реакции и регенерацией свободного фермента. Как правило, общая необходимая энергия активации определяется тем количеством энергии, которое требуется для превращения ЕS в ЕР. Этот шаг и определяет скорость всей реакции.
6.1.6.2. Молекулярные механизмы ферментативного катализа
Ферменты обладают субстратной специфичностью и специфичностью действия. Степень субстратной специфичности различается для отдельных ферментов. Некоторые гидролазы, например, относительно неспецифичны, они гидролизуют различные субстраты; другие относительно специфичны д ля определенных группировок молекул. Так, α-глюкозидазы гидролизуют в различных субстратах α-глюкозидные, но не β-глюкозидные связи. Многие ферменты в высшей мере субстратспецифичны. Бросается в глаза часто наблюдаемое распознавание ими стереоизомеров. Под этим понимают различающуюся (в основном, очень значительно) скорость оборота метаболитов, которые отличаются друг от друга всего лишь пространственным расположением замещающих групп (например, цис-транс-изомеров, или оптических изомеров, которые ведут себя, как картина и ее зеркальное отражение, — их называют энантиомерами).
Субстратспецифичность основана на определенном специфическом соответствии между субстратом и каталитически активным сайтом фермента, активным центром. В простейшем случае субстрат и каталитический центр подходят друг к другу как ключ и замок. Эта метафора, введенная Эмилем Фишером уже в 1890 г., не берет, однако, в расчет тот факт, что часто связывание ферментом субстрата — процесс динамический, в ходе которого конформация фермента и субстрата изменяется. Этот процесс, постулированный Э. Кошландом в 1958 г., называется индуцированным соответствием (англ. induced fit). Активный центр фермента часто формируется лишь после того, как состоялось связывание субстрата и индуцированное им изменение конформации, как в случае фосфоглицераткиназы (рис. 6.8).
Рис. 6.8. Обусловленное связыванием с субстратом изменение конформации (индуцированное соответствие), схематично представленное на примере фосфоглицераткиназы. После связывания субстратов АДФ и 1,3-ди- фосфоглицерата конформация белка коренным образом изменяется, при этом оба домена фермента смыкаются над связанными субстратами при одновременном исключении воды (в середине). В возникшем свободном от воды реакционном пространстве происходит перенос фосфатной группы. После восстановления «открытой» конформации фермента продукты реакции диффундируют из каталитического центра. Представлен схематический срез через активный центр фермента, участники реакции показаны в приблизительно одинаковом масштабе
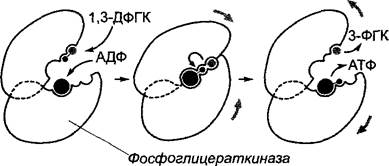
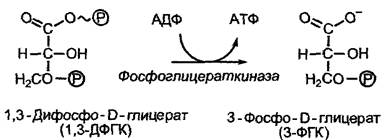
Таблица 6.4. Международная классификация энзимов: обозначение класса, кодовое число и тип катализируемой реакции
Энзимы называются, как правило, по экспериментально обнаруженной реакции, однако катализируют в клетке при определенных условиях обратную реакцию (пример: шикиматдегидрогенеза, см. 6.13.2, рис. 6.107). Классификация составлена по правилам, устаноаленным комиссией по энзимам (англ. Enzyme Commission), IUB (international Union of Biochemistry, Международный союз по биохимии). Каждый энзим получает 4-значное кодовое число: например, Е.С.1.1.1.25 является кодом энзима шикиматдегидрогеназы (см. рис. 6.107):

1. Оксидоредуктазы (редакция окисления-восстановления)
1.1. Действует на > СН—ОН
1.2. Действует на > С—О
1.3. Действует на > СН=СН—
1.4. Действует на > СН—NН2
1.5. Действует на > СН—NН—
1.6. Действует на НАДН; НАДФН
2. Трансферазы (перенос функциональных групп)
2.1. С1-группы
2.2. Альдегидные или кето-группы
2.3. Ацильные группы
2.4. Гликозильные группы
2.5. Алкильные или арильные группы (кроме метильных)
2.6. N-содержашие группы
2.7. Р-содержашие группы
2.8. S-содержащие группы
3. Гидролазы (гидролитические реакции)
3.1. Сложноэфирные соединения
3.2. Гликозидные соединения
3.3. Эфирные соединения
3.4. Пептидные соединения
3.5. Другие С—N-соединения
3.6. Кислотно-ангидридные соединения
4. Диазы (разрушают С—С, С—О, С—N и другие соединения)
5. Изомеразы (изомеризация, т.е. внутримолекулярные изменения)
5.1. Рацемазы, эпимеразы
5.2. Цис-транс-изомеразы
5.3. Внутримолекулярные оксидоредуктазы
6. Ли газы (синтетазы*) (ковалентное соединение между двумя молекулами с одновременным расщеплением АТФ)
* Энзимы анаболических реакций, которые протекают без расщепления АТФ, называют синтазами.
Фосфоглицераткиназа, фермент гликолиза (см. 6.10.1), связывает 1,3-дифосфоглицерат и аденозиндифосфат (АДФ) и катализирует перенос остатка фосфорной кислоты с карбоксильной группы 1,3-дифосфоглицерата на АДФ с образованием АТФ и 3-фосфоглицерата. При этом разрывается одна ангидридная связь (в 1,3- дифосфоглицерате) и образуется другая (в АТФ). Эта реакция в водной среде была бы совершенно невозможна, так как энергетически гидролиз был бы более предпочтителен. Решение проблемы заключается в том, что связывание АДФ и 1,3-дифосфоглицерата создает индуцированное соответствие, при котором оба домена фермента (см. рис. 6.8) складываются над связанными субстратами, исключая воду. Лишь при этом формируется активный центр и делается возможным перенос фосфатной группы. По окончании катализа «открытая» конформация вновь восстанавливается и продукты реакции диссоциируют от фермента.
Помимо субстратспецифичности ферменты обладают специфичностью действия. Это значит, что биокатализатор катализирует лишь одну из большей части многочисленных термодинамически возможных реакций превращений субстрата. Что касается работающих при этом механизмов, существует лишь относительно немного типов реакций, на основе которых и создана систематическая номенклатура ферментов (табл. 6.4).
Название фермента (для расщепляющих субстрат ферментов) выбирают таким образом, что окончание -аза добавляется к названию субстрата: например, протеиназа — для расщепляющих белок, амилаза — для гидролизующих крахмал (лат. amylum) и липаза — для расщепляющих жир (греч. lipos) ферментов. Кроме этого, в употреблении были и остаются исторически сложившиеся названия, как, например, пепсин, каталаза. Единая систематическая международная обязательная классификация и наименование всех известных энзимов были предложены Международной комиссией по ферментам (International Enzyme Commission); при этом каждый фермент получил свой классификационный номер (Е.С.), которым он однозначно идентифицируется (см. табл. 6.4). Так как систематические наименования отчасти сильно усложнены, в употреблении помимо них остаются также более краткие тривиальные названия.
В то время как у ряда ферментов каталитически активен белок как таковой, другим требуются дополнительные вещества (кофакторы). Такими кофакторами могут быть ионы металлов (к примеру, Mg2+, Mn2+, Zn2+, Fe2+, Fe3+, Cu2+, K+), которые могут требоваться для закрепления субстрата на молекуле фермента или же могут участвовать в самой реакции в качестве каталитической группы. Если в качестве кофакторов требуются органические соединения,
то их называют коэнзимами. Если коэнзим соединен с белковой частью фермента так прочно, что отделяется от него лишь с трудом (например, не выделяется из комплекса с ферментом путем диализа), то его обозначают как простетическую группу (греч. ргоsthetos — добавленный). Так, например, в цитохромах гем ковалентно связан с белком (см. 6.4.6; 6.10.3.3). Весь комплекс фермента с кофактором обозначают как холоэнзим, а саму белковую компоненту (энзиматически неактивную) ферментов — как апоэнзим.
Если кофакторы (как, например, в окислительно-восстановительных реакциях) используются стехиометрически к субстрату, то их можно обозначить так же, как косубстраты.
Катализируемая ферментом реакция превращения субстрата в его продукт протекает согласно представленной на рис. 6.7 общей схеме. Для введения основных понятий кинетики ферментативных реакций реакцию можно представить в упрощенном виде
![]()
При этом k+1 k-1 и k +2 означают константы скоростей отдельных частичных реакций (в обратных секундах). В упрощенной модели примем, что обратная реакция Е + Р —> ESпротекает пренебрежимо медленно (k2 ≈ 0) и распад комплекса энзим-субстрат (фермент-субстратного комплекса) (ES) на фермент + продукт (Р) протекает намного медленнее, чем обратная реакция ES —> Е + S (k+2⋘ k-1). Поэтому шагом, определяющим скорость всей реакции, будет превращение ES —> Е + Р, так как самая медленная частная реакция определяет и скорость общей реакции. Скорость превращения субстрата в его продукт при этих условиях задается уравнением
![]()
Максимальная скорость будет достигаться, когда весь фермент присутствует в форме комплекса с субстратом:
![]()
При скорости, равной половине максимальной (1/2Vmax), в наличии имеется столько же свободного фермента, сколько и фермент-субстратного комплекса ЕS: [ЕS] = [Е]. При равновесии формирование фермент-субстратного комплекса описывается выражением
![]()
и таким образом, после преобразования, принимая, что к+2⋘ к-1
![]()
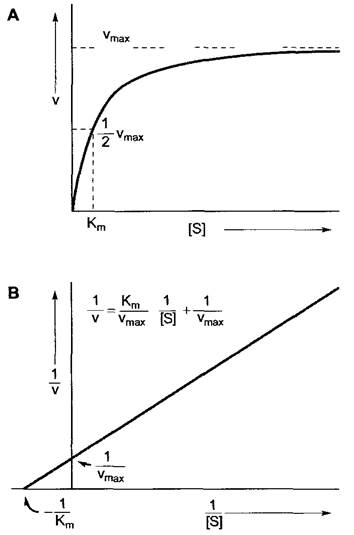
Отношение к-1/к+1 называют константой Михаэлиса—Ментен (Кm). Она может быть определена как концентрация субстрата, при которой достигается [ЕS] = [Е] (см. выше). Кm, таким образом, указывает на концентрацию субстрата, при которой достигается точно половина максимальной скорости реакции.
Так как по графику зависимости v относительно [S] (рис. 6.9, А) ни Vmax (и таким образом 1/2Vmах) > ни соответствующую концентрацию субстрата нельзя определить с достаточной точностью, Кm лучше определять после линейной трансформации графика, представленного на рис. 6.9, А, что достигается отображением той же зависимости в обратных координатах (1/v относительно 1/[S] — диаграмма по Лайнвиверу— Бёрку; рис. 6.9, В). Для данного фермента, данного субстрата и при данной температуре Кт есть константа и выражается она в моль л-1. Значения Кm можно определить также и для кофакторов.
Рис. 6.9. Зависимость (А) скорости (v) от концентрации субстрата [S] катализируемой ферментом реакции в соответствии с моделью Михаэлиса—Ментен (см уравнения 6.25 и 6 26) (по Lineweaver — Burk). В — vmax и Km можно точнее определить после построения графика в обратных координатах
6.1.6.4. Влияние среды на активность ферментов
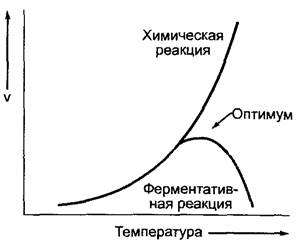
Ферментативная активность определяется в значительной степени температурой, значением pH и ионным составом среды. Эти факторы оказывают воздействие на структуру белка-фермента. Зависимость скорости реакции от температуры имеет вид кривой с оптимумом (рис. 6.10). Оптимум действия различается для отдельных ферментов и часто лежит между 30 и 50 °С. До достижения оптимума скорость реакции удваивается или утраивается при повышении температуры на каждые 10 °С. Соотношение между скоростями реакции VT!!!+10/VT называется значением Q10. Для катализируемых ферментами реакций значение Q10=2 — 3. При температурах выше оптимума в большинстве случаев активность очень быстро падает, что объясняется термической денатурацией белка-фермента; из-за сильного возрастания энтропии денатурация оказывается более предпочтительной реакцией. Отдельные ферменты очень термостабильны. Так, например, рибонуклеаза и пероксидаза могут выдержать даже кипячение. Замораживание переносится большинством ферментов без вреда, по этой причине растворы ферментов обычно хранят в замороженном состоянии.
Рис. 6.10. Зависимость скорости (V) от температуры некатализируемой (или катализируемой небелковым катализатором) и катализируемой ферментом химической реакции. Температурные оптимумы большинства ферментов лежат между 30 и 50°С
Часто в связывании, в каталитическом превращении субстрата или в построении конформации белка-фермента задействованы ионизируемые группы субстрата или фермента1. Тогда активность фермента зависит от значения pH окружающей среды. Зависимость ферментативной активности от pH может быть ярко выражена, а рН- оптимумы различных ферментов или для одного фермента, но для разных субстратов, могут лежать при весьма различных значениях pH.
1 В белках обычно представлены ионизируемые группы, способные обратимо присоединять ионы Н+: -СООН, -NH2 и др. Степень ионизации зависит от концентрации Н+ в растворе, т. е. от pH. — Примеч. ред.
Так, например, Н+-АТФаза плазмалеммы имеет рН-оптимум 6,5, аргиназа для аргинина — рН-оптимум 9,7; фумараза имеет два рН-оптимума: с фумаратом в качестве субстрата — 6,5, а с малатом — 8,5; кислые фосфатазы обладают оптимумом pH в районе 5. В широкой области значений pH активность остается неизменной, например, у инвертазы, которая расщепляет электрически нейтральный субстрат (сахароза) и для которой известны как внеклеточные, так и внутриклеточные изозимы (см. 6.8.4.)
Так как многочисленные ферменты клетки имеют различные pH-оптимумы и в отдельных компартментах значения pH различаются, изменения значений pH в клетке существенно влияют на обмен веществ. Ионный потенциал («ионная сила») также может влиять на белки-ферменты. Ионный потенциал оказывает влияние в числе прочего на конформацию ферментов через их степень гидратации.
В конце концов активность фермента прямо зависит также от концентрации субстрата и, если фермент работает в комплексе с отделяемым кофактором, — от концентрации кофактора (см. рис. 6.9). При разветвлениях путей метаболизма от концентрации общего субстрата может зависеть, какое направление будет предпочтительным. При ограниченном количестве субстрата будет преимущественно работать тот фермент, который имеет более низкую константу Михаэлиса—Ментен.
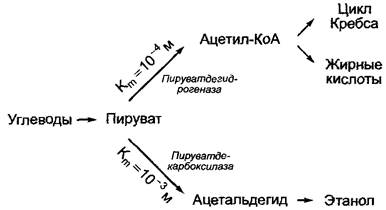
У организмов, которые способны к спиртовому брожению (например, дрожжей), пируват может либо декарбоксилироваться пиру- ватдекарбоксилазой с образованием ацетальдегида, либо за счет действия пируватдегидрогеназы окислительно декарбоксилироваться. При низких концентрациях пирувата из-за более низкого значения константы Михаэлиса—Ментен пируватдегидрогеназы протекает преимущественно образование ацетил-коэнзима А, в то время как при более высоких концентрациях пирувата на первый план выступает образование ацетата (рис. 6.11).
Рис. 6.11. Направление потоков метаболитов при разветвлении обмена веществ зависит от концентрации общего субстрата и значения Кт конкурирующих ферментов в точке разветвления. Чем ниже концентрация субстрата, тем более предпочтительно протекает реакция через путь, катализируемый ферментом с наиболее низким значением Кm. Как пример представлен метаболизм пирувата за счет действия пируватдегидрогеназы или пируватдекарбоксилазы
6.1.7. Регуляция ферментативной активности
Подобно всем белкам, ферменты постоянно синтезируются и распадаются в клетке. При этом скорость, с которой протекает данная реакция в клетке, в первую очередь контролируется через количество фермента. Этот процесс может иметь важное значение для адаптации к изменившимся метаболическим потребностям обмена веществ, но идет слишком медленно, чтобы тонко регулировать метаболизм. Поэтому наряду с контролем количества фермента существуют многочисленные эффективные и главным образом обратимые процессы, которые служат для прямого контроля ферментативной активности и дают клетке возможность быстро и мобильно приспосабливать обмен веществ к изменившимся потребностям.
6.1.7.1. Контроль количества фермента
Количество какого-либо белка в клетке является результирующей скорости его синтеза и распада. Скорость синтеза белка зависит от транскрипционной активности кодирующего гена (см. 7.2.2) и посттранскрипционных процессов, которые включаются соответственно после синтеза мРНК. Последние определяют, например, стабильность мРНК и, таким образом, вместе с синтезом мРНК ее количество в клетке. Дальнейшие механизмы регуляции касаются процессов трансляции, т.е. влияют на перевод кода нуклеиновых кислот в соответствующую линейную последовательность аминокислот (см. 7.3.1.2) и, когда это необходимо, — процессинга первично образованного полипептида до зрелого, энзиматически активного белка. Так, к примеру, расщепляющая жир липаза в прорастающих семенах Ricinus высвобождается из белка-предшественника за счет действия протеиназы.
Во многих случаях в растении присутствует несколько изозимов. Изозимами называют ферменты, которые, хотя и осуществляют одну и ту же реакцию, но от
личаются по своим химическим свойствам (например, по своей изоэлектрической точке — см. 1.3.1 или по pH-оптимуму — см. 6.1.6.4). Изозимы часто являются продуктами различных генов, однако могут быть также продуктами посттранскрипционных процессов, которые ведут к различно модифицированным вариантам. В ферментах с четвертичной структурой (см. 1.3.2.3) число изозимов может быть далее увеличено за счет образования комплексов между изоформами протомеров (гетероолигомеризация). Генные семейства, кодирующие изозимы, имеют то преимущество, что каждый ген через отдельный промотор может специфическим образом регулировать свою транскрипцию (см. 7.2.2.3). Это позволяет организму поддержать специфику распределения ферментативной активности, например, в зависимости от компартмента, ткани или стадии развития, или же реагировать на многообразие различных природных раздражителей, причем, например, при повышении потребности индуцируемый фермент может синтезироваться в дополнение к постоянно активному конститутивному ферменту. Наконец, изозимы могут различаться по механизмам контроля ферментативной активности (см. рис. 6.14).
Контроль количества фермента для первичного метаболизма выражен в меньшей степени, чем для ферментов, обеспечивающих специальные функции, и тех, которые начинают синтезироваться лишь при соответствующих запросах или образуются в больших, чем ранее, количествах. Примерами этого могут служить защитные реакции растения против вредителей или возбудителей болезней (см. гл. 9). Одним из ферментов первичного метаболизма, регулируемого в том числе на уровне количества, является нитратредуктаза. Синтез этого фермента индуцируется нитратом (NO-3) и репрессируется аммонием (NH+4). В отличие от него, конститутивные ферменты, ответственные за поддержание «основного хозяйства» клетки, или энзимы «домашнего хозяйства» (англ, housekeeping enzymes), преимущественно регулируются через механизмы контроля активности.
6.1.7.2. Контроль ферментативной активности
Ферменты могут изменять свою активность за счет обратимой ковалентной модификации или же нековалентного взаимодействия с регуляторными молекулами (модуляторами). Частыми ковалентными модификациями являются фосфорилирование и дефосфорилирование, которые катализируются специфическими протеин- киназами или фосфопротеинфосфатазами. Как правило, донором фосфата является АТФ (рис. 6.12, А), а серин, треонин, тирозин или гистидин выступают как акцепторные аминокислоты при регуляторном фосфорилировании. Так, фосфоенолпируваткарбоксилаза фосфорилируется по специфическому остатку серина и при этом активируется; пируватортофосфатдикина- за инактивируется за счет фосфорилиро- вания по специфическому треониновому остатку (см. 6.5.8, 6.5.9). Многочисленные ферменты, например, находящиеся в хлоропластах и митохондриях, подлежат ре- докс-контролю через дитиолдисульфид- ную модификацию (см. рис. 6.71). Тиоре- доксины, небольшие белки с молекулярной массой около 12 кДа, которые присутствуют в растении в нескольких изоформах в цитоплазме, митохондриях и пластидах, поставляют при этом многочисленные восстановительные эквиваленты. Примерами ферментов, которые регулируются через дитиол-дисульфидную модификацию за счет тиоредоксина, являются ферменты цикла Кальвина, частности фруктозо-1, 6-бисфосфатфосфатаза и фосфорибулокиназа (см. 6.5.3). Так как фотосинтетический транспорт электронов протекает только на свету и при этом образуется восстановленный тиоредоксин, редоксконтроль ферментов цикла Кальвина служит для адаптации фотосинтетической СO2-фикси- рующей активности к смене дня и ночи (см. 6.5.5).
Рис. 6.12. Регуляция активности ферментов за счет обратимых ковалентных модификаций: А — фосфорилирование—дефосфорилирование; В — регуляция глутаминсинтетазы Е coli путем аденилирования—деаденилирования
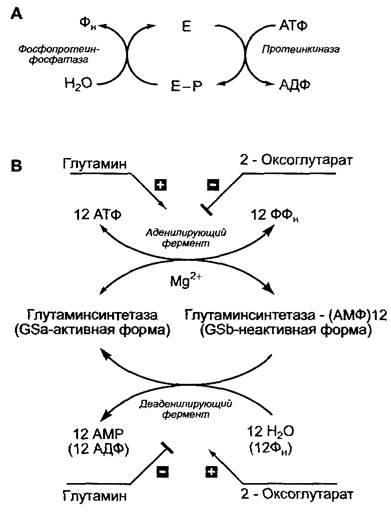
У Escherichia coli глутаминсинтетаза может присутствовать в активной или неактивной форме; в последнем случае 12 субъединиц фермента связаны с 12 молекулами аденозинмонофосфата (АМФ). Фермент, катализирующий реакцию аденилирования, т.е. инактивирующий глу- таминсинтетазу, ингибируется 2-оксоглутаратом и активируется глутамином, в то время как деаденилирующий фермент, который вновь переводит глутаминсинтетазу в активную форму, наоборот, ингибируется глутамином и стимулируется 2-оксоглутаратом (рис. 6.12, Б). Система приводит в действие автоматическую саморегуляцию синтеза глутамина из 2-оксоглута- рата: если в наличии имеется много 2-оксоглу- тарата и мало глутамина, синтез этой аминокислоты возрастает; в обратном случае — останавливается.
Изменение активности ферментов через нековалентное взаимодействие может происходить или в самом каталитическом центре, или в удалении от него. Если в каталитическом центре связывается молекула, структурно родственная его субстрату, которая тем не менее не может быть метаболизирована, то говорят о конкурентном ингибировании, так как за счет избытка субстрата ингибитор может быть вновь удален. Мера конкурентного ингибирования зависит, таким образом, от соотношения между концентрациями ингибитора и субстрата. Кинетически конкурентный ингибитор отличается тем, что Vmах реакции в присутствии ингибитора не изменяется, однако значение Кт возрастает. Если продукт ферментативной реакции действует как конкурентный ингибитор к субстрату этой реакции, то говорят об ингибировании продуктом. Этот механизм гарантирует использование лишь такого количества субстрата, которое может быть далее переработано в последующих реакциях, так что предотвращается накопление невостребованных метаболических интермедиатов.
Конкурентные ингибиторы (рис 6 13) могут быть в высшей степени эффективны. Это в особенности относится к случаю, если они представляют собой структурные аналоги переходного состояния активированного субстрата. Тогда даже при большом избытке субстрата они лишь с трyдом удаляются из фермента. Конкyрентными ингибиторами являются сульфаниламиды — бактерицидные препараты, действие которых основано на том, что они конкурентно блокируют включение близкой по структуре парааминобензойной кислоты в фолиевую кислоту — соединение необходимое для синтеза пуриновых нуклеотидов (см 6 14) Так как сам человек не синтезирует фолиевой кислоты, но получает ее как витамин с пищей, сульфаниламиды не ингибируют его обмен веществ 1
1 Отсюда следует, что применение сульфаниламидных препаратов вместе с витаминами (фолиевой кислотой) лишено смысла бактерии при этом не гибнут — Примеч. ред.
Рис. 6.13. Примеры конкурентных ингибиторов и соответствующих субстратов ферментов (см текст)
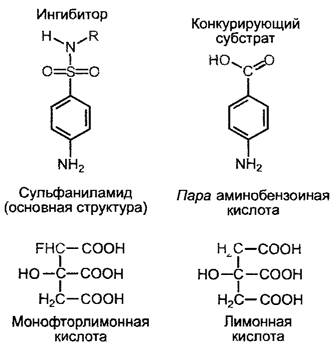
Монофторуксусная кислота CH2F— СООН представляет собой ядовитое вещество, синтезирующееся в листьях высокотоксичного для пастбищного скота южноафриканского растения Dichapetalum cymosum (сем Dichapetalaceae) Монофторацетат может связываться вместо ацетильного остатка с коэнзимом А и далее может быть перенесен ферментом цитратсинтетазой вместо ацетильного остатка на оксалоацетат (цикл лимонной кислоты см 6 10 3 2), в результате чего образуется монофторцитрат. Это соединение является в высшей степени действенным конкурентным ингибитором аконитазы — фермента, перерабатывающего цитрат в цикле лимонной кислоты. В растении Dichapetalum соответствующий токсический эффект, вероятно, предотвращается за счет того, что токсин не достигает места своего специфического действия, т. е. митохондрий, но остается заключенным в другом компартменте — вакуоли.
Рис. 6.14. Тонкая регуляция параллельных метаболических путей по механизму отрицательной обратной связи конечными продуктами аллостерически регулируемых изозимов. Накопление продукта Z ингибирует его собственное образование, однако не влияет на метаболические пути к продуктам X и Y начинающиеся от того же интермедиата В
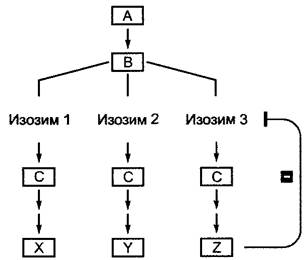
Для аллостерически регулируемых ферментов связывание модулятора влияет на изменение конформации фермента (греч. alios — другой, stereos — форма), за счет чего каталитический центр или инактивируется (модулятор действует как аллостерический ингибитор), или активируется (модулятор действует как аллостерический активатор). Если модулятор тождествен субстрату, то говорят о гомотропных ферментах, а если отличен от субстрата — о гетеротропных ферментах. Аллостерический контроль широко распространен в обмене веществ и весьма эффективен. Часто этим способом регулируются ключевые ферменты метаболических путей, которые в основном катализируют первый шаг цепи реакций и ингибируются накапливающимся конечным продуктом всей цепи. Это отрицательная обратная связь (англ. feedback inhibition) очень экономична, так как гарантирует, что поток метаболитов через сложные метаболические пути регулируется в соответствии с потребностями. Если концентрация конечного продукта в клетке понижается, то аллостерический ингибитор освобождается из фермента, и переработка субстрата возобновляется либо активируется. В связи с наличием изозимов в разветвленных реакционных цепях удается раздельная регуляция каждой частной цепи ее конечным продуктом, который регулирует соответствующий изозим (первый вслед за разветвлением) по механизму отрицательной обратной связи (рис. 6.14; см. также рис. 6.108; 6.112). Положительная обратная связь присутствует тогда, когда модулятор активирует аллостерический фермент.
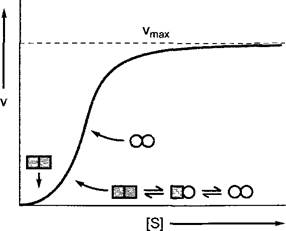
Аллостерически регулируемые ферменты, как правило, состоят из нескольких субъединиц, которые в своей активности зависят одна от другой. Такое поведение называется кооперативностью. Осуществляемое при связывании модулятора (для гомотропных ферментов — субстрата в одном из каталитических центров; для гете- ротропных ферментов — регулятора на другом участке комплекса) изменение конформации передается прочим субъединицам и изменяет (в основном, повышает) сродство остальных каталитических центров к субстрату. Из-за этого кривые насыщения субстратом аллостерически регулируемых ферментов имеют сигмоидную форму (рис. 6.15). В соответствии с этим ферменты перерабатывают субстрат эффективно лишь начиная с определенной пороговой концентрации, сверх которой уже малые изменения концентрации субстрата приводят к решительным изменениям в скорости его превращения.
Рис. 6.15. Влияние концентрации субстрата [S] на скорость [V] реакции, катализируемой гомотропным, аллостерическим ферментом. Аллостерические ферменты часто состоят из нескольких субъединиц (показаны две), если субстрат отсутствует, находятся в малоактивной форме (квадраты). Связывание субстрата одной из субъединиц индуцирует переход всех субъединиц в высокоактивную форму (круги). Из-за этого скорость реакции при низких концентрациях субстрата повышается вначале медленно, а затем экспоненциально. Как только фермент переведен в активированную форму, зависимость скорости реакции от [S] приближается к кинетике Михаэлиса — Ментен
6.1.7.3. Регуляция за счет объединения ферментов в мультиферментные комплексы или компартменты
Существенным основанием для упорядоченного, контролируемого протекания обмена веществ в клетке является объединение ферментов для определенных последовательностей реакций (реакционных циклов) в мультиферментных комплексах или целых областей метаболизма — в определенных компартментах.
В мультиферментном (мультиэнзимном) комплексе несколько ферментов объединены в сверхструктуру. За счет такой организации обеспечивается быстрое, упорядоченное превращение вещества в несколько следующих один за другим этапов. Если при этом промежуточные продукты отследить не удается, то говорят о канализировании метаболитов(англ. metbolite channeling). Мультиферментным комплексом является комплекс пируватдегид- рогеназы (см.6.10.3.1) или комплекс синтетазы жирных кислот дрожжей (см. 6.11.1). Специфический ферментный комплекс мицелиальных грибов объединяет пять ферментативных активностей пути биосинтеза ароматических аминокислот, за которые у Escherichiacoli отвечают отдельные ферменты, в одном-единственном пентафунк- циональном полипептиде (см. 6.13.2).
Объединение целых групп ферментов, кофакторов и метаболитов в реакционных пространствах, которые отделены от своего окружения метаболическими барьерами (т.е. компартментах, таких, как цитоплазма, хлоропласт, митохондрии), имеет решающее значение для упорядоченного протекания обмена веществ клетки и его контроля. Обмен метаболитами между компартментами осуществляется главным образом через специфические переносчики — транспортеры (транслокаторы — англ. carrier) (см. рис. 6.4), активность которых также регулируется различными способами.
Разобранные в этом разделе принципы биоэнергетики, ферментативного катализа и регуляции будут полезны для понимания многообразных функций растения, которые представлены в следующих главах.