Фізіологія людини - Вільям Ф. Ґанонґ 2002
Дихання
Регулювання дихання
Хімічне регулювання дихання
Хімічні регулювальні механізми пристосовують вентиляцію легень у такому напрямі, що у нормі альвеолярний РСО2 є сталим, впливи надлишку Н+ у крові подолані, і РО2 підвищується, якщо його зниження досягає потенційно небезпечного рівня. Дихальний хвилинний об’єм пропорційний до рівня обміну речовин, проте зв’язок між обміном речовин і диханням визначений СО2, а не О2. Рецептори у каротидних та аортальних клубочках подразнювані підвищенням РСО2 або концентрацією Н+ в артеріальній крові чи зниженням РО2. Після денервації каротидних хеморецепторів відповідь на зниження РО2 неможлива. Предомінантним впливом гіпоксії після денервації каротидних клубочків є безпосередня депресія дихального центру. Відповідь на зміни концентрації Н+ в артеріальній крові у межах pH 7,3-7,5 також нівельовані, хоча більші зміни впливають частково. З іншого боку, відповідь на зміни артеріального РСО2, виникають, однак повільніше; він зменшується не більше ніж на 30-35%.
Таблиця 36-1. Подразники, що впливають на респіраторний центр

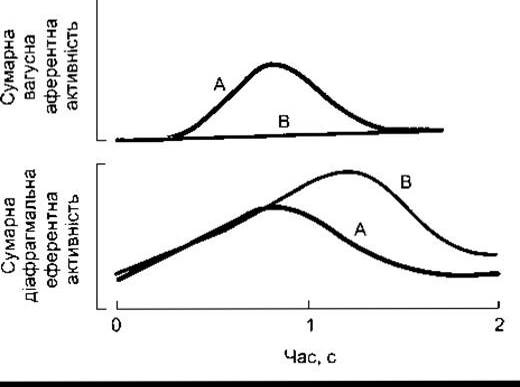
Рис. 36-3. Графічний запис двох варіантів дихання з рецепторів розтягу легень: з (А) та без (В) зворотного зв’язку впливу вагусної активності. Зверніть увагу, що рівень зростання активності діафрагмального нерва на діафрагму є незмінним, однак імпульсація триваліша без вагусного впливу.
Каротидні та аортальні клубочки
Каротидні клубочки розташовані у місці каротидної біфуркації з кожного боку, і звичайно два або більше аортальні клубочки - біля дуги аорти (рис. 36-4). Кожен каротидний або аортальний клубочок (гломус) містить острівці двох типів клітин: тип І або тип II, оточені фенестрованими синусоїдними гемокапілярами. Біля типу І, або гломусних клітин, близько є кубкоподібні закінчення аферентних нервів (рис. 36-5). Гломусні клітини подібні до хроматофінних клітин надниркових залоз, у них є щільні гранули, що містять катехоламіни, які вивільняються під впливом гіпоксії або ціанідів (див. нижче). Клітини стимулює гіпоксія і головний трансмітер, що з’являється, є дофамін, який збуджує нервові закінчення через О2-рецептори. Тип II - це гліоподібні клітини, кожна з яких оточена чотирма або шістьма клітинами типу І. Функції клітин типу II остаточно не з’ясовані.
Поза капсулою кожного клубочка нервові волокна мають мієлінову оболонку. Діаметр цих волокон 2-5 мкм, швидкість проходження через них - 7-12 м/с. Аферентні волокна від каротидних клубочків піднімаються до довгастого мозку через каротидний синус і язикогорлові нерви, а волокна від аортальних клубочків - через блукаючі нерви. Під час записування досліджень, за яких один з каротидних клубочків був ізольований і перфузований, в аферентних нервових волокнах, які працювали, простежувалося ступінчасте збільшення імпульсного потоку, якщо РО2 у перфузованій крові знижувався (рис. 36-6) або РСО2 підвищувався.
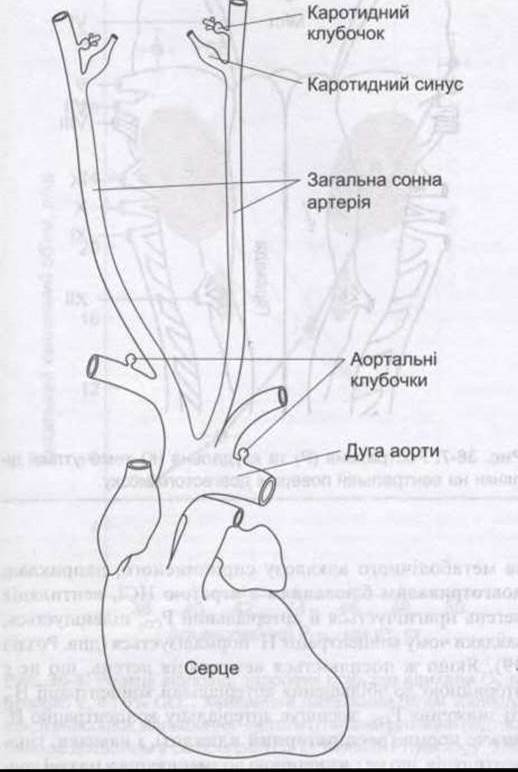
Рис. 36-4. Розташування каротидних та аортальних клубочків
Гломусні клітини типу І мають О2-чутливі К+-канали, у яких швидкість проходження зменшується пропорційно до ступеня гіпоксії, якій вони піддаються. Це зменшує витікання К+, деполяризує клітину і спричинює надходження Са2+ головно через L-тип Са2+-каналів. Надходження Са2+ є початковим моментом для утворення потенціалів дії та вивільнення трансмітера з послідовним збудженням аферентних нервових закінчень. Гладкі м’язи легеневих артерій містять подібні О2-чутливі К+-канали, які започатковують вазоконстрикцію, зумовлену гіпоксією (див. Розділ 37). Це є відмінним від загальних артерій, які містять АТФ-залежні К+-канали, що сприяють більшому витіканню К+ в разі гіпоксії і послідовно зумовлюють вазодилатацію замість вазоконстрикції. Кровоплин у кожному 2 мг каротидному клубочку становить близько 0,04 мл/хв, або 2000 мл/100 г тканини/хв, що можна зіставити з аналогічним кровоплином у 100 г/хв і становитиме 54 мл у головному мозку і 420 мл у нирках (див. табл. 32-1). Оскільки кровоплин через одиницю тканини відповідає нормі, то О2, клітини потребують більше, ніж окремо розчиненого О2. Тому подразнення рецепторів не відбувається за таких станів, як анемія або отруєння карбон монооксидом, коли кількість розчиненого кисню в крові, що досягає рецепторів, є нормальною, а вміст О2 у сполуках суттєво зменшений. Подразнення рецепторів виникає, коли артеріальний Ро2, низький або за наявності судинного стазу; кількість О2, що надходить до рецепторів за 1 хв, зменшується. Сильне подразнення також зумовлюють препарати, зокрема, такі як ціанід, які блокують утилізацію О2 на тканинному рівні. У достатніх дозах нікотин і лобелін активують хеморецептори. Також зафіксовано, що інфузійне введення К+ збільшує рівень імпульсації в аферентних волокнах хеморецепторів, тому рівень K+ у плазмі збільшується під час фізичного навантаження, збільшення може відповідати за спричинену навантаженням гіпервентиляцію.
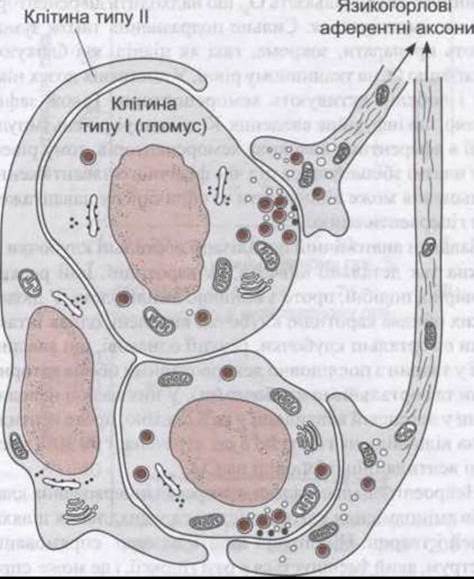
Рис. 36-5. Організація каротидного тільця. Клітини типу І (гломус) містять катехоламіни. Після впливу гіпоксії вони вивільняють ці катехоламіни, які подразнюють у каротидному синусі кубкоподібні закінчення нервових волокон, що є у складі язикогорлового нерва. Гліоподібні клітини типу II оточують клітини типу І і, ймовірно, виконують функцію підтримки.
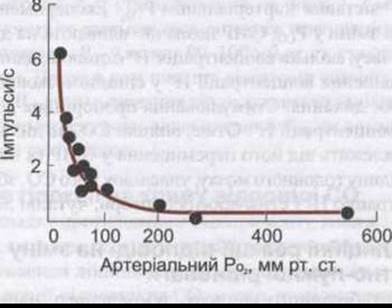
Рис. 36-6. Зміни рівня імпульсації в окремому аферентному волокні, що проходять від каротидного клубочка зі зниженням РО2 (з дозволу S Sampson).
Завдяки анатомічній локалізації аортальні клубочки не можна так детально вивчати, як каротидні. Їхні реакції, ймовірно, подібні, проте з меншою амплітудою. У людей, в яких обидва каротидні клубочки видалені, однак інтактними є аортальні клубочки, реакції однакові, що важливо (як і у тварин з послідовно денервованими обома каторидними та аортальними клубочками). У них наявні невеликі зміни у легеневій вентиляції у разі спокою, проте вентиляційна відповідь на гіпоксію буде втрачена, і на 30% зменшені вентиляційні відповіді на СО2.
Нейроепітеліальні тільця, створені з іннервованих кластерів аміновмісних клітин, містяться у дихальних шляхах людей і тварин. Ці клітини мають назовні спрямований К+-струм, який зменшується в разі гіпоксії, і це може спричинювати деполяризацію. Отже, функція цих чутливих до гіпоксії клітин повністю не з’ясована, оскільки, як описано вище, видалення одного з каротидних клубочків зумовлює зникнення респіраторної відповіді на гіпоксію.
Хеморецептори у стовбурі головного мозку
Хеморецептори, що започатковують гіпервентиляцію, спричинену збільшенням артеріального РСО2 після денервації каротидних та аортальних клубочків, містяться у довгастому мозку, тому їх називають хеморецепторами довгастого мозку. Вони відокремлені від дорсальних та вентральних дихальних нейронів і локалізовані на вентральній поверхні довгастого мозку (рис. 36-7).
Моніторинг концентрації Н+ у СМР, а також в інтерстиційній рідині головного мозку, виконують хеморецептори; СО2 швидко проникає через мембрани, зокрема, гематоенцефалічний бар’єр, тоді як Н+ і НСО3 - повільно. Потрапляючи у головний мозок та СМР, СО2 швидко гідратується. Унаслідок дисоціації Н2СО3 збільшується місцева концентрація Н+. В інтерстиційній рідині головного мозку концентрація Н+ зіставна з артеріальним РСО2. Експериментально створені зміни у РСО2 СМР незначно вливають на дихання стільки часу, скільки концентрація Н+ є сталою, однак будь- яке збільшення концентрації Н+ у спинномозковій рідині стимулює дихання. Стимулювання пропорційне до збільшення концентрації Н+. Отже, впливи СО2 на дихання головно залежать від його переміщення у СМР та інтерстиційну рідину головного мозку, унаслідок чого СО2 збільшує концентрацію Н+ і стимулює рецептори, чутливі до Н+.
Вентиляційні реакції-відповіді на зміну кислотно-лужної рівноваги
За метаболічного ацидозу, зумовленого, наприклад, нагромадженням у кровообігу кетоацетонових продуктів у разі цукрового діабету, відбувається респіраторне стимулювання (дихання Куссмауля; див. Розділ 39). Натомість за метаболічного алкалозу спричиненого, наприклад, довготривалим блюванням з втратою НСl, вентиляція легень пригнічується й артеріальний РСО2 підвищується, завдяки чому концентрація Н+ нормалізується (див. Розділ 39). Якщо ж посилюється вентиляція легень, що не є вторинною до збільшення артеріальної концентрації Н+, то зниження РСО2 зменшує артеріальну концентрацію Н+ нижче норми (респіраторний алкалоз), і навпаки, гіповентиляція, що не є вторинною до зменшення у плазмі концентрації Н+, спричинює респіраторний ацидоз.
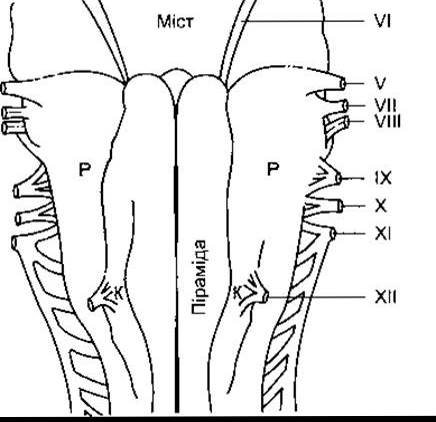
Рис. 36-7. Ростральна (Р) та каудальна (К) хемочутливі ділянки на вентральній поверхні довгастого мозку.
Вентиляційні реакції-відповіді на СО2
У нормі артеріальний PCО2 є на рівні 40 мм рт. ст. Якщо внаслідок пришвидшення обміну речовин у тканинах його рівень підвищується, то вентиляція легень посилюється, і рівень легеневої екскреції СО2 збільшуватиметься, доки артеріальний РСО2 не стане нормальним. Процес механізму цього зворотного зв’язку утримує екскрецію та утворення СО2 у рівновазі.
Коли вдихають газову суміш, що містить СО2, то підвищуються альвеолярний та артеріальний РСО2, і стимулювання вентиляції легень розпочинається, як тільки кров, що вміщує більшу кількість СО2, досягне довгастого мозку. Елімінація СО2 збільшується, і альвеолярний РСО2 знижується ДО норми. Ось чому порівняно великі Прирости РСО2 у повітрі, яке вдихають (наприклад, 15 мм рт. ст.) зумовлюють незначне підвищення альвеолярного РСО2 (наприклад, 3 мм рт. ст.). Отже, РСО2 не нормалізується, і нова рівновага настане, коли альвеолярний РСО2 незначно підвищиться, а гіпервентиляція триватиме стільки, скільки вдихатимуть СО2. Істинне лінійне співвідношення між дихальним хвилинним об’ємом і альвеолярним РСO2 зображене на рис. 36-8. Проте є обмеження такої лінійності. Коли РСО2 газової суміші, яку видихають, близький до альвеолярного РСО2, то елімінація СО2 утруднена. Якщо ж вміст СО2 у газовій суміші, яку видихають, понад 7%, то альвеолярний та артеріальний РСО2 починають збільшуватись раптово незважаючи на гіпервентиляцію. Наслідком стає нагромадження СО2 в організмі (гіперкапнія), що пригнічує центральну нервову систему, у тім числі дихальний центр, і спричинює біль голови, неспокій і, зрештою, кому (СО2- наркоз).
,
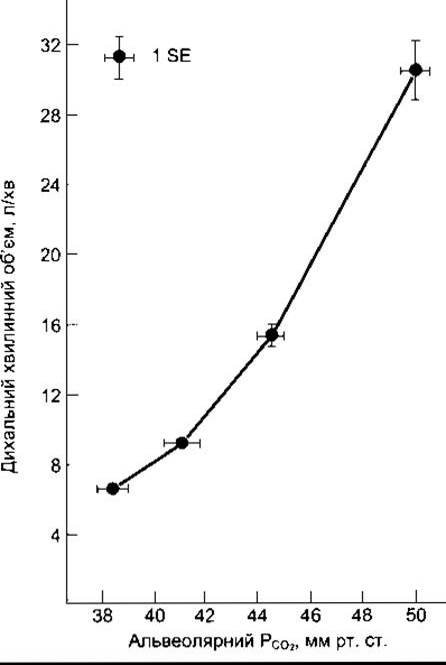
Рис. 36-8. Реакції відповіді здорових осіб, що вдихали 02 та близько 2, 4 і 6% СО2. Хвилинний дихальний об’єм збільшується внаслідок збільшення частоти і глибини дихання (відтворено за дозволом з Lambertsen CJ in: Medical Physiology, 13th ed. Mountcastle VB [editor]. Mosby, 1974).
Вентиляційні реакції-відповіді на втрату кисню
Коли вміст О2 у повітрі, яке вдихають, зменшується, то це спричинює збільшення хвилинного об’єму дихання. Стимулювання є незначне, доки РCО2 у повітрі, яке вдихають, становить понад 60 мм рт. ст., і посилюється тільки у разі нижчих значень РCО2 (рис. 36-9). Отже, будь-яке зниження артеріального РCО2 до 100 мм рт. ст. зумовлює збільшення імпульсації в нервах від каротидних та аортальних хеморецепторів. Є два погляди на те, чому у здорових осіб у нормі таке збільшення імпульсного потоку не спричинює збільшення легеневої вентиляції доти, доки РО2 не стане меншим 60 мм рт. ст. Оскільки Нb є слабшою кислотою, ніж НbО2 (див. Розділ 35), то простежується незначне збільшення концентрації Н+ в артеріальній крові, коли РО2 знижується, і гемоглобін стає менш насиченим О2. Унаслідок зменшення концентрації Н+ виникає схильність до гальмування дихання. Додатково будь-яке збільшення легеневої вентиляції, що знижує альвеолярний РСО2, також зумовлює пригнічення дихання. Тому стимулювальні впливи гіпоксії на легеневу вентиляцію не повністю з’ясовані, доки вони не стають вираженими, і достатньо знехтувати протилежними до врівноваження пригнічувальними впливами на зменшення артеріальної концентрації Н+ і РСО2.
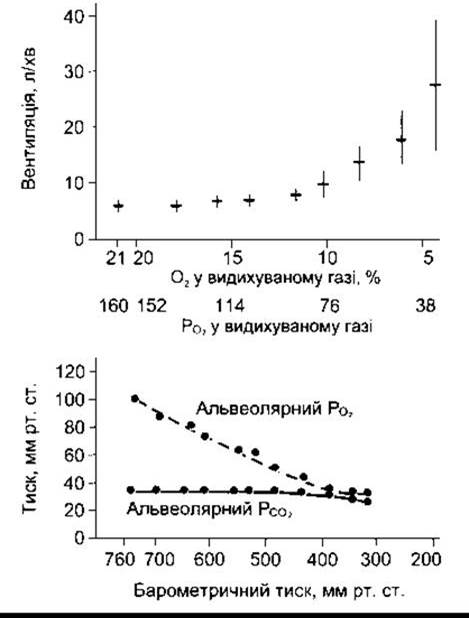
Рис. 36-9. Угорі: середні значення хвилинного дихального об’єму після впливу газових сумішей з різним вмістом О2. Горизонтальна лінія у кожному випадку відповідає середньому значенню, вертикальна шкала - одному стандартному відхиленню. Внизу: значення альвеолярного PО2 і РСО2 під час вдихання повітря та за різних барометричних тисків. Два графіки вирівнюються, так що РО2 у газових сумішах, які вдихають (верхній графік), узгоджується з РО2 для різних барометричних тисків (нижній графік) (дані надані RH Kellogg).
Вплив зниження альвеолярного РO2 за сталого альвеолярного РСО2 на вентиляцію зображено на рис. 36-10. Якщо альвеолярний РСО2 стабілізований на рівні 2-3 мм рт. ст. понад норму, то взаємовідношення між вентиляцією легень і альвеолярним РО2 у межах 90-100 мм рт. ст. стають протилежними. Проте в разі нижчих від норми значень альвеолярного PCО2 стимулювання гіпоксією вентиляції легень не відбуватиметься, доки альвеолярний РО2 не знизиться до 60 мм рт. ст. і менше.
Вплив гіпоксії на криву відповіді СО2
У випадку протилежного експерименту, коли альвеолярний РО2 є сталим, реакції відповіді на різноманітні кількості СО2 визначені лінійними співвідношеннями (рис. 36-11). Коли реакції-відповіді на СО2 мають різні значення РО2, то нахил кривої відповіді змінюється: збільшується зі зниженням альвеолярного РО2. Іншими словами, гіпоксія робить особу чутливішою до підвищення артеріального РСО2, хоча рівень альвеолярного РСО2, за якого криві на рис. 36-11 перетинаються, є незмінним. У нормі в осіб таке порогове значення є нижчим від нормального альвеолярного РСО2, і це свідчить, що у нормі є дуже мале, однак відчутне «зміщення СО2» для дихального центру.
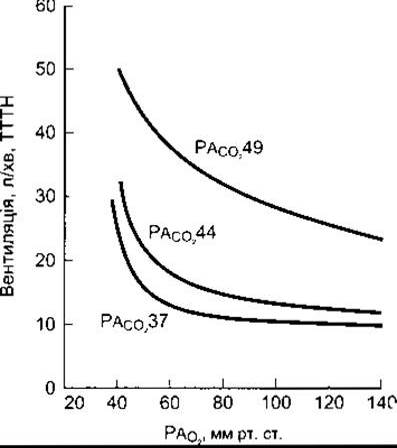
Рис. 36-10. Вентиляція легень для різних значень альвеолярного РО2 за стійкого утримування РСО2 на рівні 49, 44, або 37 мм рт. ст. (дані надані Loeschke НН і Gertz КН).
Вплив Н+ на відповідь СО2
Стимулювальні впливи Н+ і СО2 на дихання виявляються, подібно як СО2 і О2, що повністю взаємовідносно. У разі метаболічного ацидозу крива відповіді СО2 подібна до кривих на рис. 36-11, за винятком того, що ці криві зміщені ліворуч. Іншими словами, дихальне стимулювання однаково виникає і за нижчих артеріальних рівнів РСО2. Обчислено, що зміщення кривої відповіді СО2 на 0,8 мм рт. ст. ліворуч на кожний наномоль збільшуватиме артеріальний вміст Н+. Понад 40% вентиляційної відповіді на СО2 скасовуватиметься, якщо завадити збільшенню артеріального Н+, створеного СО2. Як зазначено вище, решта 60%, можливо, відповідають за вплив СО2 на концентрацію Н+ у спинномозковій або інтерстиційній рідині головного мозку.
Затримка дихання
Дихання можна свідомо затримувати на деякий час, проте остаточно довільне регулювання обмежене і його не беруть до уваги. Межа, коли дихання не можна більше утримувати затриманим, називають точкою розриву. Вона залежить від підвищення артеріального РСО2 і зниження РО2. Особи можуть затримувати дихання довше після видалення каротидних клубочків. Дихання 100% киснем перед затримкою дихання зумовлює підвищення альвеолярного РО2, завдяки чому точка розриву настає пізніше. Це однаково справджується в разі гіпервентиляції повітрям кімнати, оскільки СО2 вивітрюється назовні й артеріальний РСО2 є низьким на початку. Рефлекторно або завдяки механічним факторам розпочинається вплив на точку розриву, тому особи, які затримують дихання якомога довше, а потім вдихають газову суміш з низьким вмістом О2 і високим СО2, можуть затримати дихання на додаткових 20 с або більше. Психологічні фактори також відіграють роль, і особи, які можуть утримувати дихання довше, коли вони говорять, своїм виступом свідчать про добрий стан, ніж коли ні.
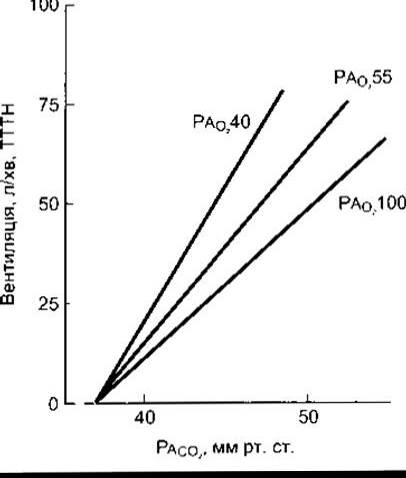
Рис. 36-11. Графічне лінійне зображення («віяло») відповідей СО2 у разі різних незмінних рівнів альвеолярного РО2.
Гормональні впливи на дихання
Вентиляція легень збільшується під час лютеїнової фази місячного циклу, а також вагітності (див. Розділ 23). Експерименти з тваринами свідчать, що це залежить від активування естрогенозалежних прогестеронових рецепторів у гіпоталамусі. Однак фізіологічне значення такого збільшення остаточно не з’ясоване.