Молекулярная биотехнология. Принципы и применение - Глик Б., Пастернак Дж. 2002
Молекулярная биотехнология микробиологических систем
Молекулярная генетика человека
Клонирование генов заболеваний человека
Как правило, ген конкретного заболевания человека нельзя клонировать, руководствуясь каким-то заранее составленным набором экспериментальных протоколов. Выбор имеющихся в распоряжении исследователя методов и средств зависит от конкретных условий. ЕІачало поиска гена заболевания определяется имеющейся информацией о продукте данного гена. В одних случаях генный продукт бывает хорошо известен, в других можно лишь догадываться, что он собой представляет. Наконец, для многих на следственных заболеваний природа генного продукта вообще неизвестна. Для каждого из этих случаев разработана своя стратегия. В целом для поиска гена заболевания существует четыре подхода: функциональное, кандидатное, позиционное и позинионно-кандидатное картирование. Независимо от применяемого подхода утверждать, что данный ген ассоциирован с интересующим исследователя заболеванием, можно лишь после того, как у больных обнаружены нуклеотидные изменения в гене, не встречающиеся в том же гене у здоровых индивидов.
Выявление мутаций в генах человека
Для выявления мутаций разработан целый ряд простых и недорогих подходов, таких как анализ конформационного полиморфизма одноцепочечной ДНК (SSCP, single-strand conformational polymorphism), градиентный гель-электрофорез в денатурирующих условиях (DGGE, denaturing gradient gel electrophoresis), гетеродуплексный анализ (НА, heteroduplex analysis), химическое расщепление некомплементарных сайтов (CMC, chemical mismatch cleavage), тест на укороченный белок (PTT, protein truncation test).
Наиболее широко среди перечисленных подходов применяется SSCP. Суть метода состоит в следующем. Как можно большее (по возможности все) число экзонов исследуемого гена по отдельности амплифицируют методом ПЦР, используя в качестве матрицы ДНК больных и здоровых индивидов. Каждая пара праймеров выбирается из последовательностей, фланкирующих экзон, или из его концевых участков. Кроме того, используя данные секвенирования, выбирают праймеры для амплификации 5'-области, предшествующей первому экзону гена, 3 -области, следующей за последним экзоном, и участков, содержащих сайты сплайсинга.
ПЦР -продукты каждой реакции денатурируют, быстро охлаждают и разделяют с помощью электрофореза. Благодаря внутри цепочечному спариванию комплементарных оснований и образованию других связей денатурированная одноцепочечная молекула ДНК принимает определенную трехмерную конформацию, зависящую от ее нуклеотидной последовательности. Вследствие комплементарности две цепи одной молекулы ДНК имеют разную нуклеотидную последовательность, а поэтому принимают разную трехмерную конформацию и мигрируют при гель-электрофорезе с разной скоростью. В результате после разделения в геле наблюдаются две полосы, отвечающие разным комплементарным цепям. Если две молекулы ДНК, представляющие один и тот же участок гена, но полученные из разных источников, различаются одной парой нуклеотидов, то с большой вероятностью конформации одиночных цепей таких молекул ДНК будут различаться. Другими словами, каждая из четырех цепей будет перемещаться при гель-электрофорезе со своей скоростью (рис. 20.22). С помощью метода SSCP можно лишь локализовать нуклеотидные изменения в определенном экзоне или специфической области гена, но не определить природу мутации; такую информацию может дать лишь секвенирование Метод SSCP имеет свои ограничения: он выявляет около 90% однонуклеотидных изменений в ПЦР-продуктах длиной не более 200 п. н.
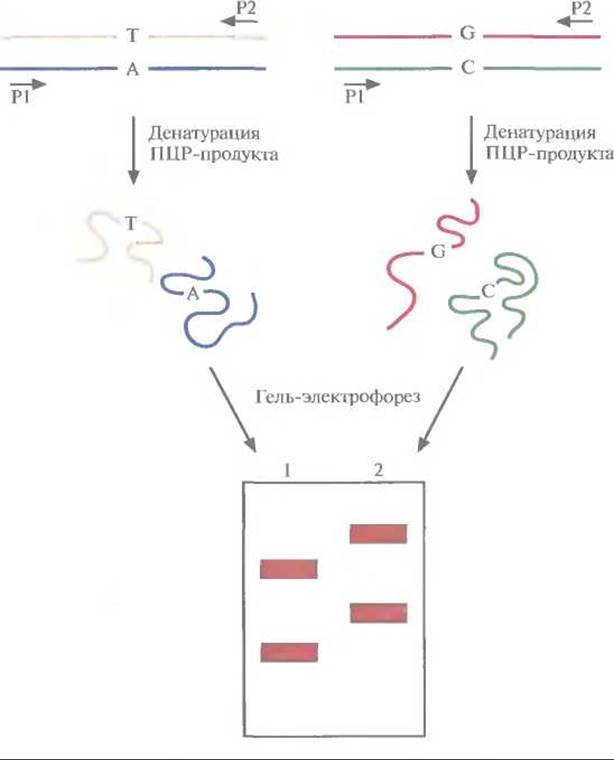
Рис. 20.22. Анализ конформационного полиморфизма одноцепочечной ДНК (SSCP). Препараты ДНК, различающихся одной парой нуклеотидов (А:Т ↔ G:C), амплифицируют ПЦР-методом с использованием одинаковых праймеров (P1, Р2). ПЦР-продукты денатурируют и разделяют с помощью гель-электрофореза на двух дорожках (1, 2). Расстояние, на которое перемещается одноцепочечная молекула ДНК, зависит от ее конформации, а последняя, в свою очередь, — от нуклеотидной последовательности. Даже если ДНК различаются лишь одним нуклеотидным сайтом, одиночные цепи могут иметь разную конформацию, а следовательно, ПЦР-продукты образуют в геле не две, а четыре полосы.
Функциональное картирование
Функциональное картирование гена начинается с определения аминокислотной последовательности белка с известной функцией, что позволяет реконструировать нуклеотидную последовательность кодирующей области соответствующего гена (гена-мишени). Основываясь на этих данных, синтезируют олигонуклеотидные зонды и проводят скрининг кДНК-библиотеки, полученной для ткани, в которой данный белок присутствует в большом количестве. Если можно полущить очищенную мРНК, с которой транслируется данный белок, то на ней как на матрице можно синтезировать полноразмерную кДНК и клонировать ее. Правильность выбора или синтеза кДНК-клона проверяют секвенированием.
Хромосомную локализацию гена-мишени определяют методом гибридизации in situ с кДНК-клоном или выбранным с его помощью геномным клоном. Для более точной локализации гена-мишени можно также провести скрининг панели монохромосомных клеточных гибридов, а затем и соответствующей делеционной панели при помощи кДНК-клона или отобранного геномного клона.
Затем для определения клонов, гибридизующихся с данным кДНК-клоном, проводят скрининг космидного контига, охватывающего хромосомный район, в котором локализован ген-мишень. Отобранные геномные клоны секвенируют и используя данные о нуклеотидной последовательности кДНК, идентифицируют экзоны, и нтроны и 5'-, 3'-фланкирующие последовательности гена. В отсутствие космидного контига, охватывающего район нужной хромосомы, который содержит ген-мишень, выделяют клон с крупной вставкой, содержащей данный район, при помощи кДНК- или геномного зонда. Из клона с крупной вставкой получают субклоны с небольшими вставками, и проводят их скрининг при помощи кДНК- зонда. Позитивные клоны секвенируют и характеризуют ген-мишень (рис. 20.23).

Рис. 20.23. Функциональное картирование. Идентификация гена для случая, когда известна аминокислотная последовательность его продукта.
Кандидатное картирование
Хотя этот подход не очень эффективен при картировании генов человека, в ряде случаев он может оказаться весьма полезным. Суть метода состоит в следующем. Анализируют симптомы генетического заболевания и на их основе пытаются понять, какого типа белок может быть с ним ассоциирован. Затем просматривают нуклеотидные последовательности всех клонированных на настоящий момент генов и выбирают ген(ы)-кандидат(ы). Основываясь на нуклеотидной последовательности гена-кандидата, вырабатывают стратегию поиска мутаций и с ее помощью пытаются установить, является ли ген-кандидат искомым геном (рис. 20.24). Принимая во внимание, что геном человека содержит очень большое число генов, а охарактеризованы лишь некоторые из них, не стоит удивляться, что правильный выбор гена случается не так уж часто. Но ценен и отрицательный результат, поскольку он позволяет исключить данный ген из числа ответственных за конкретное генетическое заболевание.
Позиционное картирование
Стратегия позиционного картирования применяется в тех случаях, когда ничего не известно о продукте гена, ответственного за наследственное заболевание, и нет никаких генов-кандидатов (рис. 20.25). В подобных случаях определяют хромосомную локализацию (позицию) гена заболевания и проводят его поиск, применяя различные инструменты и средства («охота за геном»). Благоприятной для позиционного картирования является ситуация, когда у нескольких больных встречается хромосомная перестройка типа транслокации или крупной делеции (> 10 т. п. н.). Предположив, что она затрагивает ген, ответственный за патологический фенотип, при анализе сцепления используют только один специфический район хромосомы вместо того, чтобы проводить сканирование всего генома при помощи большого числа полиморфных маркеров. После локализации гена в конкретном районе хромосомы определяют его положение более точно и идентифицируют ближайшие фланкирующие маркеры, используя мультилокусное картирование с дополнительными полиморфными зондами. Минимальное расстояние между картированными маркерными сайтами, при котором их можно разграничить, в лучшем случае составляет 1 сМ, что соответствует примерно 106 п. н. На таком участке может уместиться в среднем от 20 до 50 генов. Задача позиционного картирования состоит в том, чтобы определить, какой именно из них ответствен за данное заболевание.
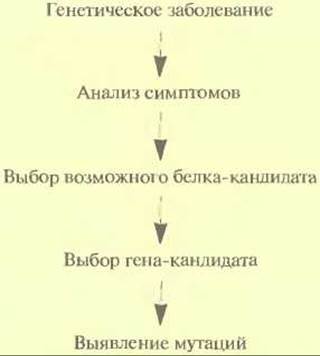
Рис. 20.24. Кандидатное картирование. Идентификация гена, основанная на анализе симптомов обусловленного им заболевания и соображений, какой из уже охарактеризованных генов может претендовать на роль искомого гена.
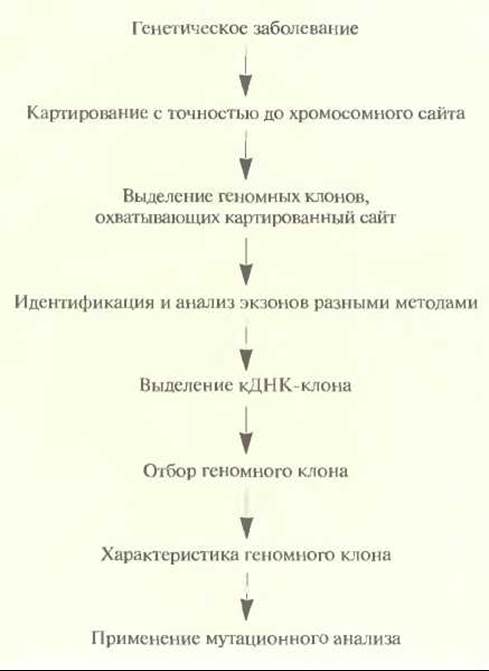
Рис. 20.25. Позиционное картирование. Идентификация гена, продукт которого неизвестен, с помощью хромосомного картирования и зондов, специфичных в отношении тесно сцепленных маркеров.
Из контига, охватывающего район хромосомы, содержащий ген заболевания, выбирают геномные клоны, которые включают фланкирующие маркеры и заключенный между ними участок ДНК. Если такой контиг отсутствует, то с помощью зондов, специфичных в отношении тесно сцепленных маркерных сайтов, проводят скрининг библиотек геномных ДНК для выявления клонов, происходящих из того района, который содержит искомый ген. Между 1986 и 1990 гг., когда метод «охоты за генами» человека еще только разрабатывался, для идентификации нужных геномных клонов использовали метод «прыжков по хромосоме» (рис. 20.26) или метод «прогулки по хромосоме» (рис. 20.27). После создания геномных библиотек, содержащих крупные фрагменты ДНК человека, и контигов эти стратегии утратили свою актуальность. Независимо от того, как именно получены нужные геномные клоны, важно знать, какие из них или из субклонов содержат экзоны. Для этого можно использовать целый ряд прямых и косвенных методов, таких как идентификация CpG-островков, межвидовой Саузерн-блоттинг, отбор гибридов, улавливание экзонов, секвенирование ДНК, компьютерный поиск.
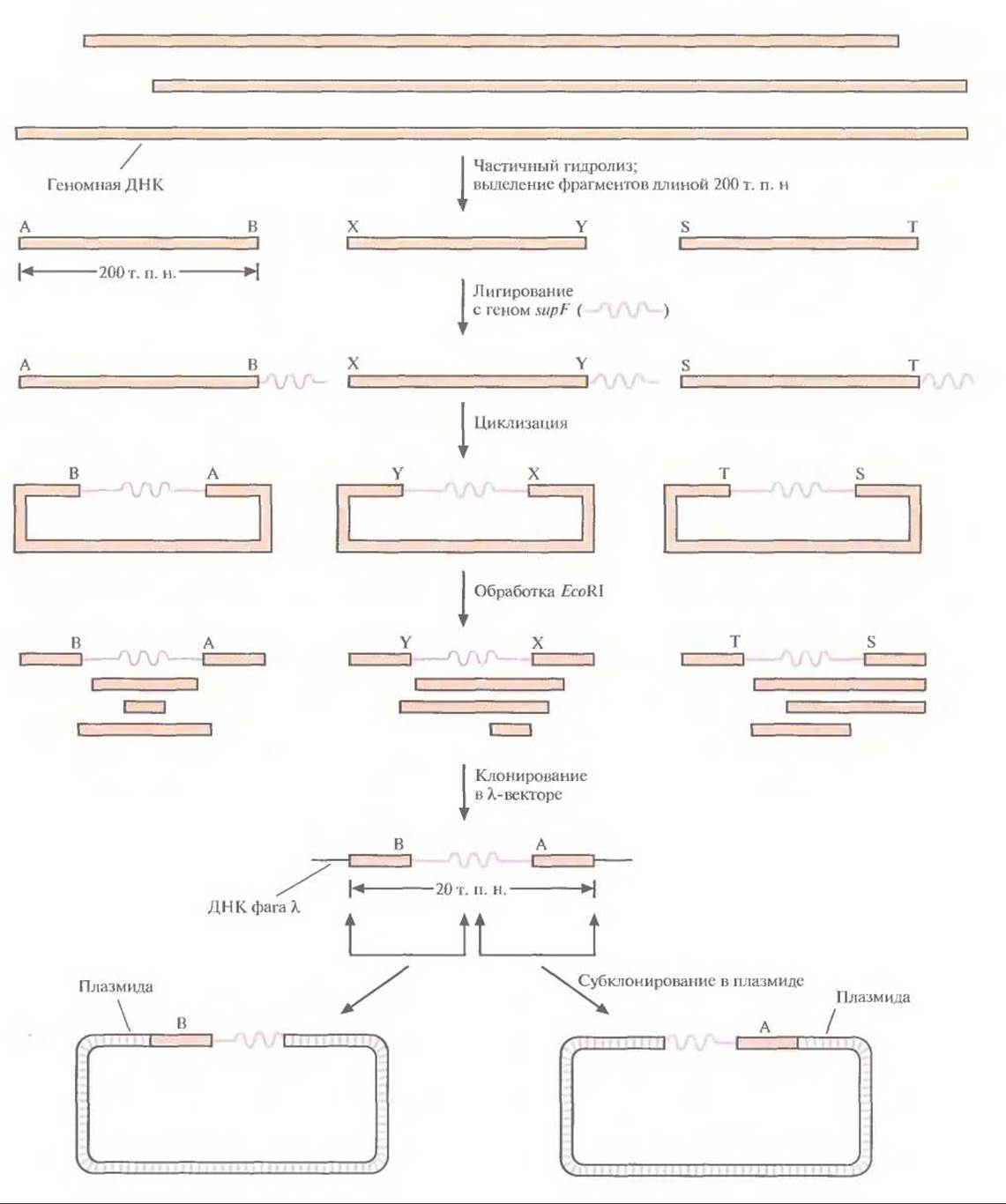
Рис. 20.26. Создание библиотеки методом «прыжков по хромосоме». Проводят частичный гидролиз геномной ДНК рестриктазой, делающей небольшое число разрывов, и выделяют фрагменты длиной примерно 200 т. п. и. Сшивают их с геном supF(~7 т. и. н.) и замыкают в кольцо. Буквами А и В, X и Y, S и Т обозначены сайты, исходно находящиеся друг от друга на расстоянии 200 т. п. н., но после циклизации фрагментов разделенные 7 т. п. н. Кольцевые молекулы, содержащие множество сайтов для ЕсоRI, обрабатывают этой рестриктазой, в результате чего среди прочих образуются фрагменты, содержащие ген supF и фланкирующие его последовательности (А и В; X и У; S и Т). Из всех фрагментов отбирают лишь те, длина которых составляет примерно 20 т. п. н., и встраивают их в вектор на основе фага λ. Векторы, несущие ген supF, будут амплифицироваться в SupF-клетках-хозяевах. Идентифицируют клон, гибридизующийся с зондом, специфичным в отношении исходной последовательности (А, X, S), затем субклонируют его; при этом та его часть, которая не гибридизуется с зондом, содержит участок ДНК (В, Y, Т), находящийся на расстоянии 200 т. п. н. от исходной последовательности.
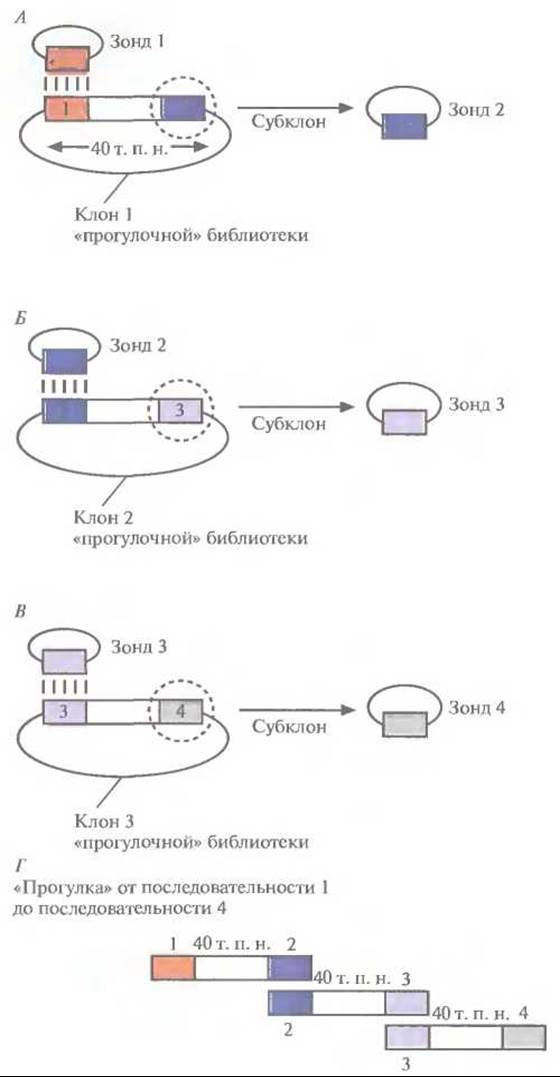
Рис. 20.27. «Прогулка по хромосоме». А. Зонд 1 гибридизуют с клонированным фрагментом ДНК длиной 40 т. п. н. После субклонирования и построения рестрикционной карты последовательность, дистальную по отношению к гибридизовавшейся, используют для создания зонда 2. Б. При помощи зонда 2 из библиотеки выбирают другой клон (отличный от клона 1) и используют последовательность, дистальную по отношению к гибридизовавшейся с ним, для создания зонда 3. Клоны 1 и 2 вместе составляют примерно 80 т. п. н. (за вычетом перекрывающегося участка — зонд 2 — между ними). В. Проводят манипуляции, аналогичные А и Б, используя зонд 3. Третий клон «прогулочной» библиотеки позволяет продвинуться по хромосоме еще на 40 т. п. н. Г. Три перекрывающихся фрагмента ДНК охватывают примерно 120 т. п. н. хромосомной ДНК. «Прогулку» по хромосоме можно совершать в двух направлениях, руководствуясь при этом рестрикционной картой.
Транскрибируемым участкам геномов позвоночных часто предшествуют кластеры нуклеотидов, богатые остатками С и G (CpG-островки). Группу CpG-островков можно идентифицировать по скоплению на рестрикционной карте сайтов для рестрицирующих эндонуклеаз EagІ, BssІІ и SacII. Если как минимум два таких сайта отделены 5—10 т. п. н. друг от друга, значит, они находятся в пределах CpG-островка. Это не гарантирует, что именно здесь находится экзон, но указывает на наличие где-то поблизости транскрибируемого гена.
Геномные клоны или субклоны можно гибридизовать по Саузерну с рестрицированной геномной ДНК различных позвоночных, например с ДНК мыши, крысы, кролика, обезьяны, коровы, цыпленка, рыбы (зооблот, межвидовой блоттинг, блоттинг «Ноев ковчег»). Положительная перекрестная гибридизация означает, что данный клон с большой вероятностью содержит кодирующие последовательности, поскольку многие экзоны в ходе эволюции не изменялись, в то время как повторяющиеся и некодирующие последовательности ДНК, в том числе и интроны, претерпели существенные изменения. Положительный зооблот означает, что клон содержит экзон(ы), однако не показывает, есть ли в нем ген искомого заболевания. Отбор гибридов позволяет быстро и с высокой эффективностью идентифицировать геномный клон, содержащий экзон(ы), и одновременно изолировать соответствующую кДНК. Отбор можно проводить разными способами. Обычно ДНК геномного клона из той области хромосомы, которая содержит ген заболевания, фиксируют на твердой подложке, проводят прегибридизацию с повторяющимися последовательностями ДНК, а затем гибридизуют с линейными векторными молекулами со вставками из кДНК-библиотеки, происходящей из ткани, вероятнее всего экспрессирующей ген-мишень. Негибридизовавшиеся векторные молекулы смывают с фильтра, а гибридизовавшиеся элюируют и амплифицируют методом ПЦР, используя праймеры из векторных последовательностей, фланкирующих кДНК-вставку (рис. 20.28). Если точно неизвестно, в какой ткани экспрессируется ген-мишень, то кДНК-библиотеки разных тканей объединяют и проводят гибридизацию с отдельными геномными клонами. ПЦР-продукт можно затем клонировать и тестировать, с тем чтобы проверить, содержит ли он кодирующую часть гена данного заболевания. Для этого можно секвенировать кДНК и провести компьютерное сравнение нуклеотидных последовательностей этой ДНК и известных генов. Если будет получена высокая степень гомологии, можно сделать определенные выводы о том, какого типа белок кодирует данная кДНК, и если этот белок таков, что его с высокой вероятностью можно считать продуктом гена-мишени, то данный(е) клон(ы) секвенируют и идентифицируют экзоны, интроны и 5'-, 3'- фланкирующие области. Альтернативный подход состоит в поиске мутаций с целью выявления нуклеотидных различий между ДНК больных и здоровых индивидов. Если подход, основанный на определении степени гомологии нуклеотидных последовательностей, оказывается безуспешным, то секвенируют и анализируют другие гены из данной области. Реально при поиске гена-мишени для экономии времени и средств характеризуют в первом приближении сразу несколько генов, пока не найдут наиболее вероятный ген-кандидат, который исследуют детально, в том числе с помощью мутационного анализа.

Рис. 20.28. Отбор гибридов. Гибридизацией с ДНК геномного клона «отлавливают» кДНК-клон, амплифицируют его, клонируют и тестируют.
Улавливание экзонов («поимка» экзонов, экзонная амплификация) — это метод, позволяющий идентифицировать и клонировать экзоны, находящиеся в субклонах, полученных из геномных клонов (рис. 20.29). Его суть состоит в следующем. ДНК геномного клона расщепляют так, чтобы получить фрагменты длиной 1 —6 т. п. н., и клонируют эти фрагменты в специально сконструированном векторе. Сайт множественного клонирования (полилинкер) вектора расположен внутри интрона, фланкированного двумя экзонами (экзон 1 и экзон 2). Этот искусственный ген (экзон 1—интрон—экзон 2) находится под контролем сильного эукариотического промотора и может реплицироваться в Е. coli или в культуре клеток млекопитающих. После введения (трансфекции) вектора без вставки в клетку млекопитающего происходит транскрипция искусственного гена и удаление интрона из первичного транскрипта. Процессированную РНК (экзон 1—экзон 2) можно выявить, проведя ПЦР-амплификапию обратного транскрипта. Сначала с помощью обратной транскриптазы на мРНК синтезируют ДНК (синтез первой цепи). Вторую цепь синтезируют при участии праймера, комплементарного части экзона 1 первой цепи. Затем в реакционную смесь добавляют второй праймер, комплементарный части экзона 2 второй цепи ДНК, и проводят ПЦР-амплификацию. Длину ПЦР-продукта определяют с помощью гель-электрофореза.
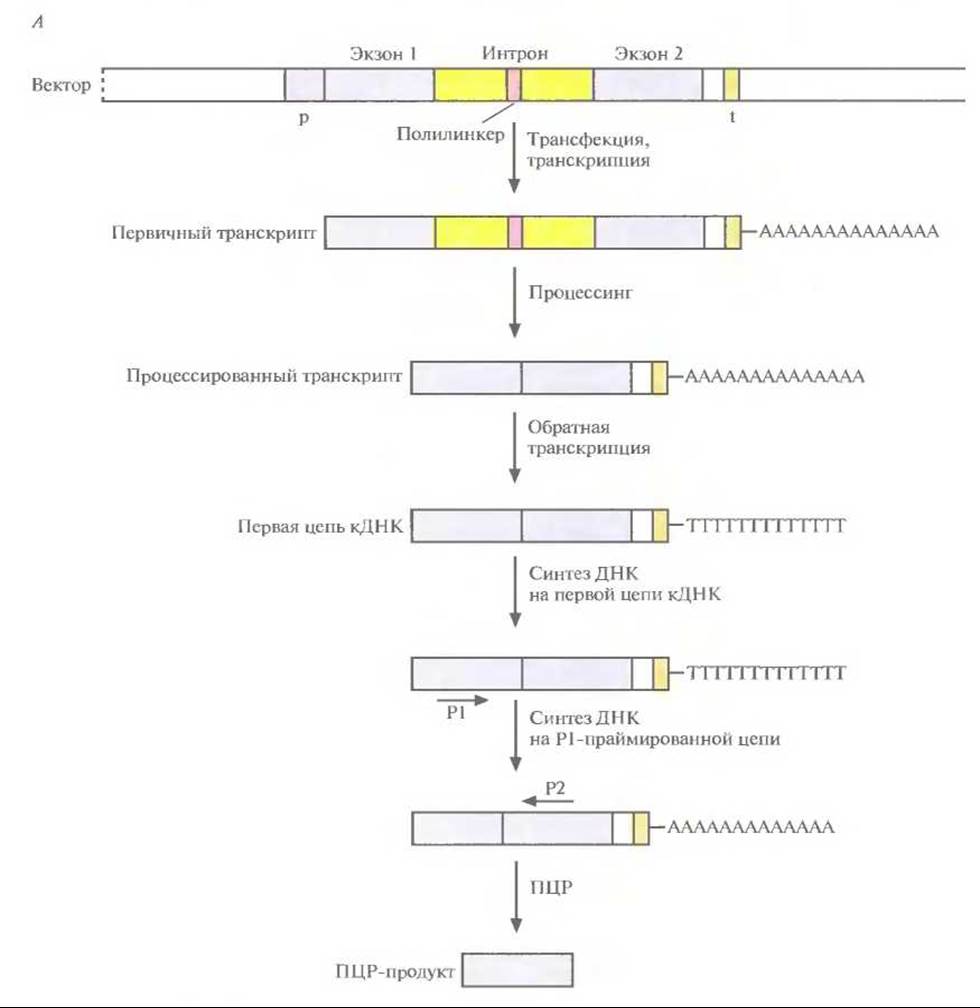
Рис. 20.29. Улавливание экзонов. А. Вектор для улавливания экзонов содержит искусственный ген, состоящий из промотора р, двух экзонов, разделенных нитроном, который несет полилинкер, и сайта терминации транскрипции t. После введения вектора в эукариотическую клетку искусственный ген транскрибируется и из первичного транскрипта удаляется интрон. Для получения ПЦР-продукта определенной длины, который содержит часть обоих экзонов, используют ПЦР-амплификацию обратного транскрипта.
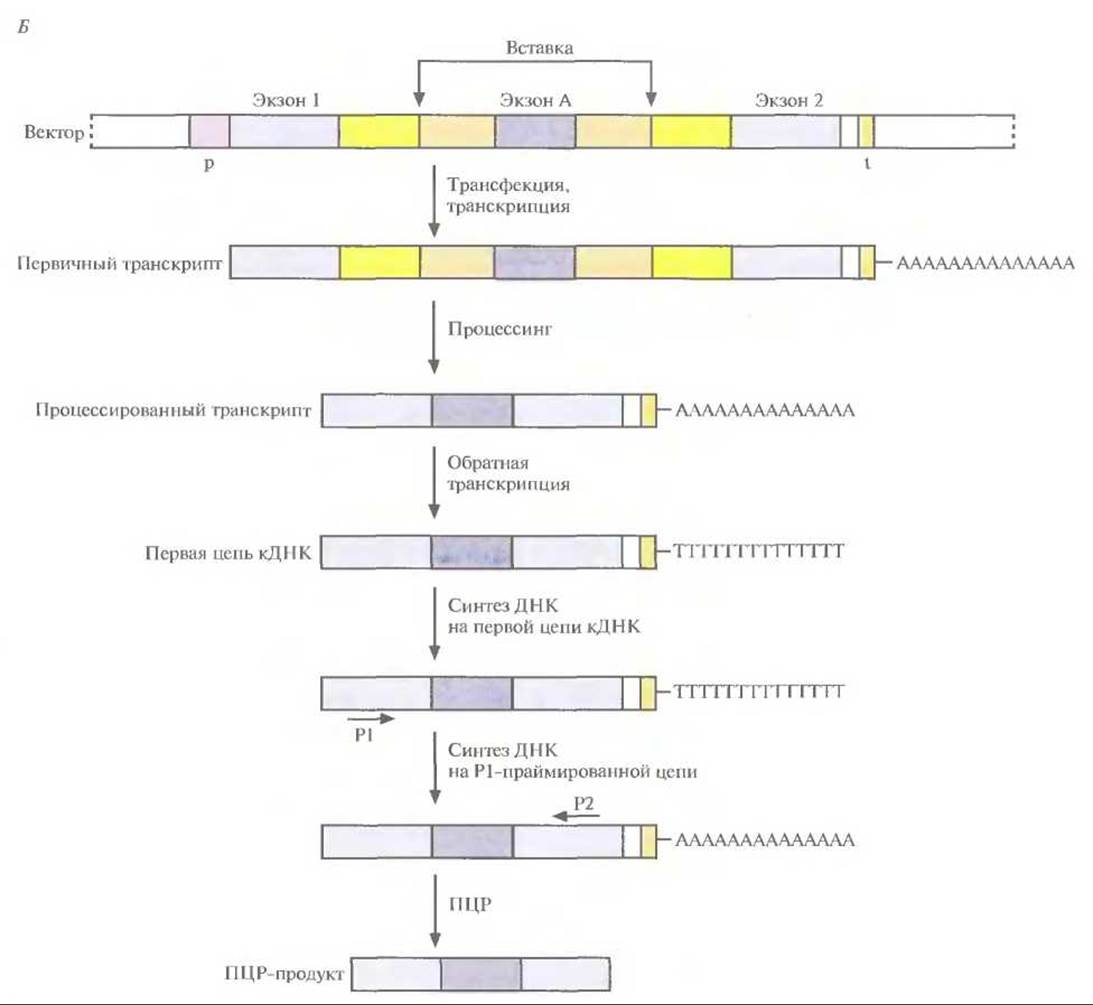
Рис. 20.29. (Продолжение) Б. В полилинкер вектора для улавливания экзонов встроен фрагмент ДИК человека, и эта конструкция введена в эукариотическую клетку (трансфекция). В данном случае экзон содержит функциональные акцепторный и донорный сайты сплайсинга. При процессинге первичного транскрипта удаляются интроны, фланкирующие экзон А, и он оказывается между экзонами 1 и 2 Длина ПЦР-продукта обратного транскрипта показывает, «пойман» ли искомый экзон между экзонами 1 и 2 и, следовательно, содержит ли его данная вставка.
Если в вектор встроить рестрикционный фрагмент, содержащий некий экзон А и фланкирующие его интроны, то после трансфекции процессированный транскрипт будет содержать три экзона: экзон 1—экзон А—экзон 2. Длина ПЦР-продукта будет больше, чем в тех случаях, когда в векторе нет вставки, когда вставка содержит экзон без функциональных сайтов сплайсинга (донорного и акцепторного) или когда вставка вообще не содержит экзона. Если во вставке присутствует более одного экзона, каждый из которых имеет функциональные сайты сплайсинга, то процессированный транскрипт будет содержать все эти экзоны.
В том случае, если в каждом праймере содержатся рестрикционные сайты, клонируют ПЦР-продукт, несущий «пойманный» экзон, и используют последний в качестве зонда для скрининга кДНК-библиотеки. Зная нуклеотидную последовательность «пойманного» экзона, предпринимают поиск гомологичных ему последовательностей в базе данных. Если есть основания полагать, что «пойманный» экзон с большой вероятностью является частью гена данного заболевания, то характеризуют и секвенируют геномные клоны, охватывающие место расположения данного гена, и исследуют образцы ДНК больных и здоровых индивидов с целью выявления мутаций. Поскольку мутации, ответственные за патологию, не всегда бывают равномерно распределены по всем экзонам, чем больше размер сканированной кодирующей области предполагаемого гена, тем больше вероятность обнаружения мутации.
Для идентификации экзонов используют различные компьютерные программы, например GRAIL (Gene Recognition and Analysis Internet Line). Они созданы исходя из некоторых характерных для экзона особенностей. Одна из них — ожидаемая нуклеотидная последовательность кодирующей области. Если лаборатория оснащена оборудованием для широкомасштабного секвенирования, можно секвенировать геномные клоны, охватывающие область расположения искомого гена, и провести компьютерную обработку полученных данных с целью выявления экзонов. Нуклеотидную последовательность предполагаемого экзона можно использовать для поиска гомологичных ей последовательностей в генной базе данных или синтезировать на ее основе олигонуклеотидный зонд для скрининга кДНК-библиотеки. Наконец, как и в случае других методов идентификации экзонов в геномных клонах, необходимо доказать, что предполагаемый экзон является частью гена-мишени.
Реализация любого проекта по позиционному картированию гена занимает много времени. За период с 1986 по 1995 г. с помощью данного подхода удалось обнаружить более 50 генов различных заболеваний человека, что можно считать большим достижением. Иногда поиск гена занимает 1—2 года, в то же время для обнаружения гена хореи Гентингтона консорциуму из нескольких исследовательских лабораторий потребовалось 10 лет. Отметим, что с клонированием все новых и новых генов и построением транскрипционных карт с высоким разрешением позиционное картирование постепенно уступает место позиционно-кандидатному.
Позиционно-кандидатное картирование
Позиционно-кандидатное картирование состоит в определении хромосомной локализации гена болезни, продукт которого неизвестен, и последующем анализе современных генетических и транскрипционных карт, с тем чтобы выявить кодирующие последовательности (гены, внутригенные EST), находящиеся в этом же районе (рис. 20.30). Весьма вероятно, что одна из этих последовательностей и окажется геном данного заболевания. Если какой-либо из генов-кандидатов охарактеризован, можно провести его мутационный анализ. Как альтернативу можно использовать «кандидатные» EST в качестве зондов, отобрать с их помощью геномный клон и секвенировать его, а затем также провести мутационный анализ. По мере детализации физических и транскрипционных карт позиционно-кандидатное картирование становится все более популярным при поиске генов различных заболеваний человека.
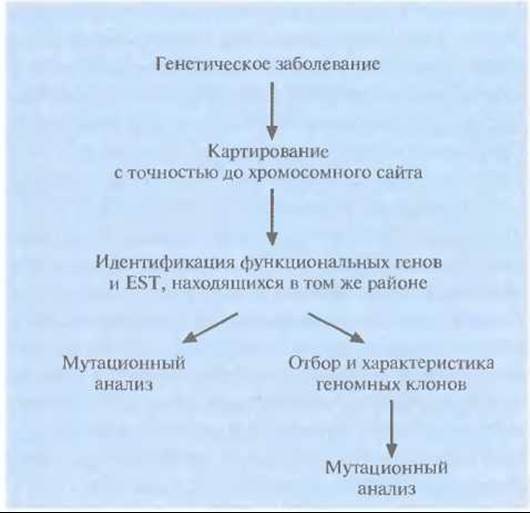
Рис. 20.30. Позиционно-кандидатное картирование. Идентификация гена заболевания в том случае, когда продукт гена неизвестен, но ген картирван в том же хромосомном районе, что и некоторые функциональные гены и EST. Из этих генов и EST отбирают «кандидатные» и определяют, какие из них соответствуют искомому гену.