Основы молекулярной биологии. Часть 2: Молекулярные генетические механизмы - А.Н. Огурцов 2011
Геномика и протеомика
Идентификация генов в геномных фрагментах ДНК
Полный геном организма содержит в себе информацию, которая определяет структуру каждого белка, синтезируемого клетками организма.
Для таких организмов как бактерии или у дрожжей, чьи геномы содержат небольшое количество нитронов и короткие межгенные участки, большинство кодирующих белки генных последовательностей может быть обнаружено методом компьютерного поиска в геноме открытых рамок считывания (open reading frames, ORFs) необходимой длины.
Открытая рамка считывания обычно определяется как участок ДНК длиной более 100 кодонов, который начинается со старт-кодона и потенциально может быть транскрибирован, а затем транслирован в полипептидную цепь. Поскольку вероятность того, что произвольный участок ДНК не содержит стоп-кодона на 100 кодонов, очень мала, то с большой долей вероятности можно ожидать, что большинство открытых рамок считывания кодируют белки.
ORF-анализ правильно идентифицирует более 90% генов в дрожжах и бактериях.
Конечно, этот метод (1) не детектирует некоторые очень короткие гены, равно как и (2) указывает на случайные длинные открытые рамки считывания, которые в действительности не являются генами.
Оба этих типа ошибок могут быть скорректированы более детальным анализом генных последовательностей и генетическим тестированием функций генов.
Например, половина из обнаруженных методом ORF-анализа генов Sacchromyces cerevisiae, была известна и ранее из исследований мутантных фенотипов. Функции некоторых белков, кодируемых остальными, обнаруженными методом ORF-анализа, предполагаемыми генами, были установлены, основываясь на их подобии с уже изученными белками других организмов.
Идентификация генов в организмах с более сложной структурой генома требует более сложного алгоритма, чем просто поиск открытых рамок считывания. На рисунке 115 для сравнения показаны фрагменты длиной 50 kb геномов дрожжей, дрозофилы и человека.
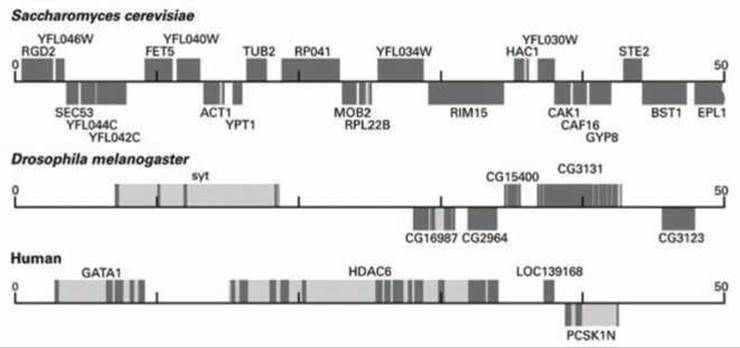
Рисунок 115 - Схема расположения генов на 50 kb участке дрожжей, плодовой мушки и человека
Гены, показанные на рисунке над сплошными линиями, транскрибируются слева направо, под линиями - справа налево. Темные участки обозначают экзоны, светло-серые - интроны.
Генные последовательности, чьи функции ещё не определены, имеют специальные обозначения: для дрожжей они начинаются с символа Y (yeast), у дрозофилы - с CG, у человека - с LOC. Остальные гены, изображенные на рисунке кодируют белки, чьи функции уже определены.
Поскольку большинство генов высших эукариот, включая человека и дрозофилу, состоят из множества относительно коротких кодирующих фрагментов (экзонов), разделенных некодирующими участками (нитронами), простое сканирование с целью поиска ORF неэффективно для поиска генов. Лучшие из алгоритмов поиска генов используют все имеющиеся данные, которые могут подсказать наличие гена в определённом месте генома.
Обычно используют такие виды информации:
1) результаты гибридизации с полной кДНК;
2) сравнение с участками кДНК длиной 200-400 пар оснований, которые называются ярлыки экспрессируемых последовательностей (EST, expressed sequence tag);
3) подгонку в соответствии с существующими моделями экзонов и нитронов;
4) подобием с генными последовательностями других организмов.
Отметим, что компьютеризация поиска генной информации в базах данных не может исключить участия человека в исследовании. Компьютер только предлагает различные варианты ответа на поставленный вопрос, решение о приемлемости и разумности предложенных вариантов принимает человек.
Объединение достижений информатики и биологии привело к появлению новых наук, таких как биоинформатика и компьютерная биология. Компьютерные биологи идентифицировали порядка 35 000 генов в геноме человека, при этом о 10 000 из этих возможных генов ещё не известно кодируют ли они в действительности белки или РНК.
В частности, очень действенным методом идентификации генов в геноме человека явилось сравнение геномных последовательностей человека и мыши. Человек и мышь в генетическом смысле являются "близкими родственниками" и имеют множество родственных генов. При этом, многие из нефункциональных ДНК-последовательностей, таких как межгенные участки или интроны, оказываются существенно различными, (поскольку они не были "оптимизированы" в ходе эволюции).
Поэтому с большой долей вероятности можно утверждать, что те участки геномов человека и мыши, которые оказались подобными, соответствуют функциональным кодирующим участкам ДНК, таким как экзоны.