МИКРОБИОЛОГИЯ БИОЛОГИЯ ПРОКАРИОТОВ ТОМ II - А. В. ПИНЕВИЧ - 2007
ГЛАВА 15. КЛЮЧЕВЫЕ БИОСИНТЕЗЫ
Синтез органического вещества является одним из основных звеньев в той цепи физико-химических процессов, из которых слагается круговорот органического вещества на земле.
В. Н. Любименко. Материя и растение (Ленинград, 1924)
Следует уточнить, что подразумевается под «ключевыми» биосинтезами, и о каких мы расскажем в этой главе.
В первую очередь, это биосинтез генеральных метаболитов. Напомним, что термин «генеральные метаболиты» обозначает интермедиаты магистральных метаболических путей, а также универсальные моноблоки, из которых образуются главнейшие биомолекулы (см. раздел 9.2). Среди них первенствуют нуклеотиды, аминокислоты, моносахара (глюкоза и рибоза) и органические кислоты (пируват и ацетил- СоА). Однако мы не станем рассматривать пути биосинтеза нуклеотидов и аминокислот, поскольку они стандартны для всех живых организмов, и их описание можно найти в любом учебнике по биохимии, а также справочниках, снабженных «метаболическими картами». Что касается разнообразных, а порой уникальных путей образования пирувата и ацетил-СоА у прокариотов, то мы уже останавливались на этом вопросе (глава 11).
В число ключевых биосинтезов входит также биосинтез биополимеров — белков, нуклеиновых кислот и полигликозидов из соответствующих генеральных метаболитов-аминокислот, нуклеотидов и моносахаров. Механизм трансляции изложен в разделе, посвященном прокариотным рибосомам (см. I том учебника), репликацию и транскрипцию мы обсудим в III томе. Наконец, основные сведения о биосинтезе полигликозидов приведены в I томе учебника, где речь шла о полимерах клеточной стенки, чехла и капсулы, а также о метаболизме запасных углеводов.
Мы специально остановимся на трех классах биомолекул, которые играют ключевую роль в функциональной организации живой клетки. Это мембранные глицеролипиды, полиизопреноиды и тетрапирролы. Сведения о биосинтезе, разнообразии и функциях этих биомолекул были впервые получены и пополняются главным образом благодаря использованию бактерий в качестве экспериментальных моделей.
Среди этих биомолекул много таких, которые не встречаются у эукариотных организмов или были приобретены ими в качестве компонента прокариотных эндосимбионтов. На биосинтезе этих ключевых биомолекул мы остановимся еще и потому, что данная тема, как правило, игнорируется в учебной литературе или освещается в самых общих чертах на основе устаревших сведений.
Во II томе учебника рассматриваются углеродная и азотная автотрофия, а также ключевые биосинтезы, однако отсутствует специальная глава, посвященная обновлению надмолекулярных структур и катаболизму экзогенных субстратов и запасных веществ.
Причина в том, что данные вопросы обсуждаются в связи с разными аспектами цитологии прокариотов (см. I том учебника) и клеточного питания (глава 11). Тем не менее, мы еще уделим внимание процессам катаболизма в III томе учебника, когда будем говорить про онтогенез и экологию прокариотов.
Начнем с глицеролипидов. Это они формируют гидрофобное ядро мембранных структур, отвечающих за важнейшие «операционные» функции прокариотной клетки, в частности за ее энергетический метаболизм.
15.1. Биосинтез глицеролипидов
Еще раз напомним о принципиальных различиях между глицеролипидами архей, с одной стороны, и глицеролипидами бактерий и эукариотов, с другой стороны (см. I том учебника).
Археи обладают тремя уникальными особенностями структуры мембранных глицеролипидов:
— гидрофобные хвосты представляют собой не терминально разветвленные или неразветвленные жирные кислоты (как в полярных липидах Bacteria и Еucarya), а регулярно разветвленные С20-) С25-) С40- или С50-полиизопреноидные спирты, в которых соседние метальные группы разделены пятью или четырьмя атомами углерода;
— глицерол имеет не sn- 1,2-конфигурацию (как в полярных липидах Bacteria и Еuarya), a sn-2,3-конфигурацию;
— гидрофобный хвост связан с глицеролом не сложной эфирной (как в большинстве полярных липидов у Bacteria и Еuсагуа), а простой эфирной связью.
Естественно, что биосинтез уникальных глицеролипидов, имеющихся у архей, осуществляется иным способом, чем у бактерий.
15.1.1. Биосинтез глицеролипидов у бактерий
Основой двойного липидного слоя бактериальных мембран служат глицеролипиды. Роль гидрофобных хвостов в них выполняют жирные кислоты. Важнейшими глицеролипидами, в частности у Е. coli, являются фосфатидилэтаноламин (70-80%), фосфатидилглицерол (15-20%) и кардиолипин (5%).
Наряду со своей основной функцией, фосфатидилглицерол используется для биосинтеза липопротеинов и периплазматических олигосахаридов (см. I том учебника).
Ряд ферментов, участвующих в биосинтезе жирных кислот, служит мишенью для антибиотиков и химиотерапевтических препаратов (см. ниже). Эти вещества используются в медицинских целях, в частности для борьбы с туберкулезом.
Биосинтез насыщенных жирных кислот. Большинство живых организмов способно самостоятельно вырабатывать длинноцепочечные жирные кислоты. Для этого они используют ферментный комплекс, состоящий из ацетил-СоА- карбоксилазы и синтазы жирных кислот. В качестве ключевого продукта образуется неразветвленная насыщенная пальмитиновая кислота (С16). Ее производными являются другие жирные кислоты, в том числе ненасыщенные, гидроксилсодержащие и с числом атомов углерода >16.
Магистральный путь биосинтеза жирных кислот был выяснен в конце 1960-х годов на примере Е. coli прежде всего благодаря пионерским исследованиям Конрада Блоха (К. Bloch, Нобелевская премия по физиологии и медицине, 1973 г.). Он осуществляется с помощью двух ферментных систем — ацетил-СоА-карбоксилазной и синтазной.
Ацетил-СоА-карбоксилаза. На исходном этапе ацетил-СоА-карбоксил аза (англ. acetyl-CoA carboxylase, ACC) осуществляет АТФ-зависимое карбоксилирование затравки-праймера — ацетил- СоА. В результате этого образуется удлинитель цепи — малонил-СоА:
СН3—CO~SCoA + HCO-3 + АТФ —> -OOC-CH2-CO~SCoA + АДФ + Ф.
У Е. coli ACC состоит из четырех типов субъединиц:
— белкового переносчика АссВ молекулярной массой 17 кДа, ковалентно связывающего карбоксибиотин (см. рис. 82);
— биотинкарбоксилазы АссС (субъединицы 33 и 49 кДа), карбоксилирующей биотин;
— карбоксилтрансферазы AccAD молекулярной массой 35 кДа, переносящей карбоксильную группу от карбоксибиотина на ацетил-СоА.
Синтаза жирных кислот. После образования малонил-СоА в дело вступает синтаза жирных кислот (англ. fatty acid synthase, FAS). Она проводит несколько раундов «конденсации», т. е. надстраивает алифатическую цепь, начиная с затравки-праймера (ацетил-СоА), за счет очередного С2-блока. Каждый раз в качестве удлинителя цепи выступает малонил-АСР. Затем происходит восстановление кетогруппы, дегидратация и восстановление двойной связи. Синтаза FAS бывает двух типов — FASI и FASII.
Синтаза FASI имеется у всех эукариотов, а также у некоторых бактерий. Она представляет собой один или два (у грибов и Corynebacterium ammoniagenes) мультифункциональных белка, содержащих несколько активных центров.
Синтаза FASII имеется у большинства бактерий. Это растворимый мультиэнзимный комплекс, компоненты которого содержат по одному активному центру и сгруппированы вокруг ацилпереносящего белка АСР (8,5 кДа). Интермедиаты пути биосинтеза жирных кислот образуют тиоэфирную связь с простетической группой АСР — 4′-фосфопантотеновой кислотой (см. рис. 93). Последняя присоединена к аминокислотному остатку Ser-36 и передается на апопротеин от коэнзима А при помощи АСР-синтазы.
Сначала ацетил-СоА: АСР-трансацилаза (32 кДа) переносит ацетильную группу от ацетил-СоА на АСР:
СН3—CO~SCoA + АСР-SH —> CH3-CO~SACP + CoASH.
Аналогичным образом, малонил-СоА: АСР-трансацилаза передает малонильную группу на АСР:
-OOC-CH2-CO~SCoA + ACP-SH —> СР-синтазы III (33 кДа) из ацетил-АСР и малонил-АСР образуется ацетоацетил-АСР. В качестве «платы» за образование четырехуглеродного скелета из активированного двухуглеродного акцептора (ацетила) и активированного трехуглеродного ацильного остатка (малонила) выделяется молекула диоксида углерода:
СН3—CO~SACP + -OOC-CH2-CO~SACP —> CH3-CO-CH2-CO~SACP + ACP-SH + CO2.
После этого β-кетоацил-АСР-редуктаза (26 кДа) превращает ацетоацетил-АСР в β-окси- бутирил-АСР:
CH3-CO-CH2-CO~SACP + НАДФН —> CH3-CHOH-CH2-CO~SACP + НАДФ.
В свою очередь, из β-оксибутирил-АСР при помощи дегидратазы (17 кДа) образуется trans- кротонил-АСР:
CH3-CHOH-CH2-CO~SACP —> CH3-CH=CH-CO~SACP + H2O.
После этого эноил-АСР-редуктаза (28 кДа), чувствительная к химиотерапевтическим препаратам диазоборину, триклозану и этионамиду, восстанавливает trans-кротонил-АСР в бутирил-АСР:
CH3-CH=CH-CO~SACP + НАДФН —> CH3-CH2-CH2-CO~SACP + НАДФ.
На следующем этапе бутирил-АСР взаимодействует с малонил-АСР, в результате чего образуется бутироацетил-АСР. В качестве «платы» за конденсацию выделяется молекула СO2. Путем восстановления кетогруппы, дегидратации окси-производного и восстановления двойной связи бутироацетил-АСР превращается в гексаноил-АСР. На следующем этапе гексаноил-АСР взаимодействует с малонил-АСР и т. д. За элонгацию цепи отвечают «конденсирующие» ферменты — β-кетоацил-синтазы I, II и III (англ. ketoacyl-ACP synthase, KAS). Они чувствительны к антибиотикам церуленину и тиолактомицину, а также к химиотерапевтическому препарату изониазиду.
За семь раундов, на каждом из которых растущая алифатическая цепь взаимодействует с очередной молекулой малонил-АСР, образуется пальмитиновая кислота (C16). На этом элонгация специфически блокируется, и ацил-ACP подвергается деацилированию:
CH3-(CH2)14-CO~SACP + Н2O —> CH3-(CH2)14-COOH + ACP — SH.
Итоговое уравнение реакции биосинтеза пальмитиновой кислоты выглядит следующим образом:
ацетил-СоА + 7 х малонил-СоА + 14НАДФН —> пальмитиновая кислота + 7СO2 + 14НАДФ + 8CoASH + 6Н2O.
Длинноцепочечные жирные кислоты с нечетным числом атомов углерода (С15) и (С17), которые характерны для многих анаэробов, образуются при помощи специфических ацетил-СоА: АСР- трансацилаз. В качестве праймера здесь используется не ацетил-СоА, а пропионил-СоА.
Некоторые бактерии, например, Butyrivibrio sp. S2, не способны синтезировать длинноцепочечные жирные кислоты. Для роста таких природных ауксотрофов необходимы готовые жирные кислоты (в свободном виде или в составе глицеролипидов).
Биосинтез ненасыщенных жирных кислот. Среди мононенасыщенных жирных кислот у бактерий преобладает cis-вакценовая кислота (Cis) с двойной связью в положении 11:12.
Двойную связь в молекулу жирной кислоты можно ввести двумя путями.
«Анаэробный» путь. Он характерен для большинства бактерий (не обязательно анаэробных) и в нем не используется кислород.
Двойная связь вводится в ходе элонгации алифатической цепи на стадии С10-продукта. В результате дегидратации в положении 2:3 образуется 2,3-trans-деканоил-АСР:
CH3-(CH2)6-CO-CH2-CO~SACP —> CH3-(CH2)6-CHOH-CH2-CO~SACP (β-окси-деканоил-АСР) —> СН3 — (СН2)6—СН=СН—CO~SACP (2,3-trans-деканоил-АСР).
Затем двойная связь восстанавливается, и биосинтез продолжается до получения насыщенной жирной кислоты с цепью той или иной длины.
Однако в том случае, когда нужно получить мононенасыщенную жирную кислоту, β-окси- деканоил-АСР превращается с помощью стереоспецифической дегидратазы/изомеразы (19 кДа) в 3,4- cis-деканоил- АСР:
CH3-(CH2)6-CHOH-CH2-CO~SACP (β-окси-деканоил-ACP) CH3-(CH2)5-CH=CH-CH2-CO~SACP (3,4-сis-деканоил-АСР).
Такой продукт не может быть восстановлен при помощи обычных редуктаз. Поэтому эноилредуктазная реакция пропускается, двойная связь остается интактной, и процесс элонгации цепи продолжается, как обычно, путем взаимодействия с новыми молекулами малонил-АСР:
CH3-(CH2)5-CH=CH-CH2-CO~SACP —>—>CH3-(CH2)5-CH=CH-(CH2)7-CO~SACP (сis-пальмитолеил-SАСР) —>—>CH3-(CH2)5-CH=CH-(CH2)9-CO~SACP (сis-вакценоил-SACP).
«Аэробный» путь. Он имеется у многих аэробных бактерий, и в нем принимает участие кислород.
В данном случае cis-двойная связь вводится в алифатическую цепь при помощи специфической мембранной десатуразы. Электроны переносятся от насыщенной жирной кислоты через НАДФН на молекулярный кислород по короткой мембранной электрон-транспортной цепи, в состав которой входят флавопротеин и цитохром b-типа. Общее уравнение реакции: O2 + 4Н+/4е- —> 2Н2O. Благодаря этому пути бактерии могут изменять степень насыщенности своих мембранных жирных кислот, уже находящихся в составе фосфолипида, т. е. пост-биосинтетически.
Бактерии редко образуют полиненасыщенные жирные кислоты, и механизм введения кратных двойных связей почти не изучен.
Биосинтез разветвленных жирных кислот. У многих бактерий встречаются терминально разветвленные жирные кислоты, обладающие изо-конфигурацией (R—СН3СН3) или антеизоконфигурацией (R—СН3С2Н5).
Их биосинтез осуществляется при помощи трансацилаз, которые используют в качестве праймера не ацетил-СоА, а короткие разветвленные эфиры коэнзима А (изобутирил-СоА, изовалерил-СоА и β-метилбутирил-СоА). Эти праймеры, в свою очередь, образуются путем дезаминирования и окислительного декарбоксилирова- ния аминокислот (соответственно валина, лейцина и изолейцина).
Магистральный путь биосинтеза фосфолипидов. Бактерии синтезируют фосфолипиды при помощи интегральных или периферических ферментов, расположенных в СМ. Поскольку активные центры взаимодействуют с растворимыми субстратами, биосинтез фосфолипидов происходит на внутренней поверхности СМ. Для образования двойного липидного слоя необходимо переместить часть вновь синтезированных молекул липида в наружный полумембранный листок. Для этого существуют специальные ферменты — флиппазы (см. I том учебника).
Первым шагом на пути биосинтеза фосфолипидов является превращение sn-глицерола в sn- глицерол-3-фосфат, которое осуществляется с помощью глицеролкиназы. Альтернативный способ получения sn-глицерол-З-фосфата — это восстановление диоксиацетон-3-фосфата НАДФН- зависимой глицерол-3-фосфат-дегидрогеназой (рис. 156).
Рис. 156. Магистральный путь биосинтеза фосфолипидов у бактерий. 1 — глицеролкинаэа; 2 — глицерол-3-фосфат-дегидрогеназа; 3 и 4 — глицерол-3-фосфат-ацилтрансферазы; 5 — ЦДФ-диацилглицеролсинтаза; 6 — фосфатидилсерин-синтаза; 7 — фосфатидилсерин-декарбоксилаза; 8 — фосфатидилхолин-синтаза; 9 — фосфатидилглицеролфосфат-синтаза; 10 — фосфатидилглицеролфосфат-фосфатаза; 11 — кардиолипинсинтаза.
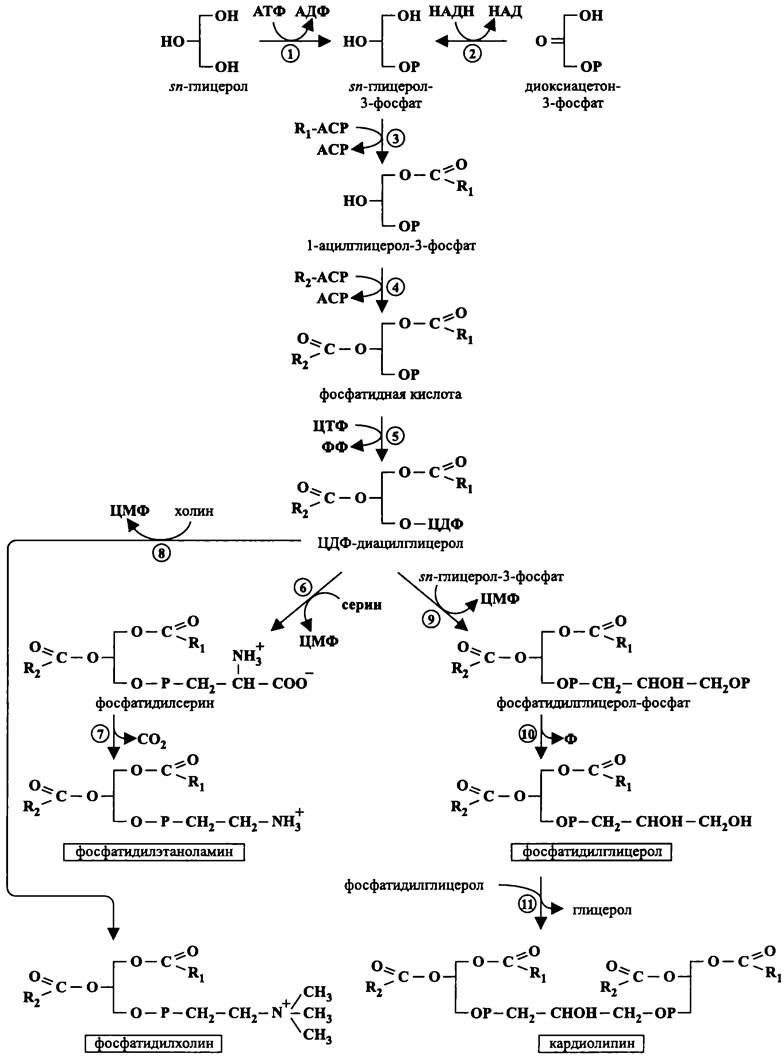
Затем глицерол-3-фосфат-ацилтрансфераза вводит в положение 1 первый остаток жирной кислоты, полученный от АСР. Другая глицерол-3-фосфат-ацилтрансфераза вводит второй остаток жирной кислоты в положение 2, в результате чего образуется ключевой интермедиат — фосфатидная кислота, которая служит общим предшественником бактериальных фосфолипидов. Обычно она присутствует в следовых количествах, поскольку соответствующая синтаза быстро превращает ее в липонуклеотид — ЦДФ-диацилглицерол.
ЦДФ-диацилглицерол дает начало двум ветвям генерального пути, которые приводят к образованию трех важнейших бактериальных глицеролипидов — цвиттерионного фосфатидилэтаноламина, анионного фосфатидилглицерола и анионного дифосфатидилдиглицерола, или кардиолипина (см. I том учебника).
Для образования фосфатидилэтаноламина требуются тоже две реакции. Вначале ЦДФ- диацилглицерол с помощью фосфатидилсерин-синтазы превращается в фосфатидилсерин. Вслед за этим фосфатидилсерин декарбоксилируется в фосфатидилэтаноламин.
Для образования фосфатидилглицерола требуются тоже две реакции. Сперва ЦДФ- диацилглицерол с помощью фосфатидилглицеролфосфат-синтазы превращается в фосфатидил- глицерол-фосфат. Продукт этой реакции дефосфорилируется с образованием фосфатидилглицерола. В свою очередь, кардиолипин синтезируется из двух молекул фосфатидилглицерола с помощью кардиолипинсинтазы. Эта реакция уникальна для бактерий (у эукариотов донором второго диацилглицерола служит ЦДФ-диацилглицерол).
Фосфатидилхолин, или лецитин, который является важнейшим мембранным фосфолипидом эукариотов, довольно редко встречается у бактерий (характерными примерами служат симбионты растений, относящиеся к родам Agrobacterium и Rhizobium). Он образуется за один этап из ЦДФ- диацилглицерола и холина с помощью фосфатидилхолин-синтазы (рис. 156).
В условиях фосфатного стресса, наступающего при фосфорном голодании, мембранные фосфолипиды замещаются (по крайней мере, частично) особыми диацил- глицеролами, роль полярных головок в которых выполняют гликозильные, в том числе сульфатированные, остатки. К их числу относятся галактоза, глюкоза и манноза, которые встречаются по отдельности или образуют разные связи друг с другом.
15.1.2. Биосинтез глицеролипидов у архей
Археи одновременно обладают глобальным малонил-СоА путем для биосинтеза жирных кислот (см. раздел 15.1.1) и глобальным мевалонатным путем для биосинтеза полиизопреноидов (см. раздел 15.2.1).
В качестве гидрофобных хвостов в мембранные липиды архей входят только по- лиизопреноидные спирты, в то время как все остальные живые организмы используют для этого жирные кислоты и очень редко — не полиизопреноидные спирты.
Хотя археи и синтезируют небольшое количество жирных кислот, последние содержатся в мембранах в свободном виде, а не в качестве компонента глицеролипидов.
Чтобы не повторяться и не погружаться в детали, мы отсылаем читателя к I тому учебника, где уже рассматривались структура и свойства липидов у архей. Остановимся только на биосинтезе.
Важнейшими и уникальными этапами биосинтеза археотных глицеролипидов являются:
— образование фитанильных гидрофобных хвостов;
— образование 3-моноалкенил-sn-глицерол-1-фосфата;
— присоединение полярных групп к преархетидной кислоте;
— образование 2,3-дифитанил-диглицерида (археола);
— образование 2,3,2',3'-дибифитанил-тетраглицерида (кальдархеола).
Помимо этого, биосинтез археотных глицеролипидов включает в себя следующие
уникальные этапы:
— образование циклопентановых колец внутри полиизопреноидной цепи;
— образование девятиатомного спирта (кальдитола).
Образование фитанильных гидрофобных хвостов. Как показали опыты по включению [1-13С]- или [2-13С]-ацетата в липидную фракцию клеток метаногенных архей Methanospirillumhungatei и Methanothrix soehngenii, фитанол образуется в мевалонатном пути (см. раздел 15.2.1). Кроме того, на примере Methanobacterium thermoautotrophicum и М. thermoformicicumустановлено, что наращивание полиизопреноидной цепи происходит при помощи классического элонгирующего фермента — пренилтрансферазы I.
Образование 3-моноалкенил-sn-глицерол-1-фосфата. 3-моноалкенил-sn- глицерол-1-фосфат, или простой моноэфир глицерола и фитанола является центральным интермедиатом на пути биосинтеза археотных глицеролипидов.
В составе моноэфира, а также образующегося из него диэфира глицерол присутствует в 2,3- sn-конфигурации (рис. 157). В результате глицеролипиды архей имеют разную стереохимию с 1,2- sn-глицеролипидами бактерий и эукариотов. Первые являются правовращающими L-изомерами, а вторые — левовращающими D-изомерами (см. I том учебника).
Рис. 157. Пути биосинтеза глицеролипидов у архей. А — экстремальные галофилы. Б — метаногены и термоацидофилы. GGОР — геранил- геранил-дифосфат, X — полярная группа.
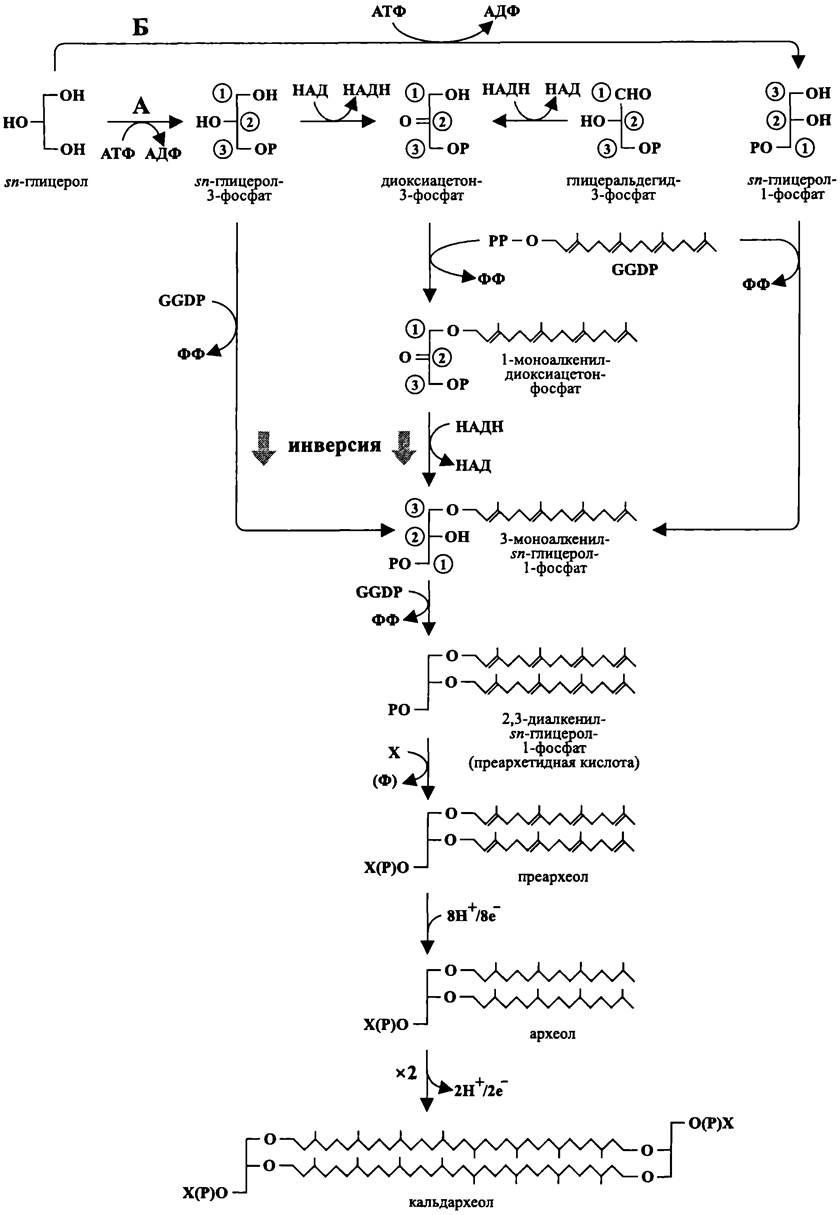
Способ, посредством которого образуется 3-моноалкенил-sn-глицерол- 1-фосфат, варьирует у разных групп архей.
Экстремально галофилъные архей (рис. 157, А). На примере Halobacterium sp. выяснено, что в результате АТФ-зависимой киназной реакции из sn-глицерола образуется sn-глицерол-З-фосфат. Затем события разворачиваются по двум сценариям.
В одном случае sn-глицерол-З-фосфат взаимодействует с геранилгеранил-дифосфатом (GGDP, см. раздел 15.2.2) с образованием 3-моноалкенил-sn-глицерол-1-фосфата. В ходе этой реакции происходит инверсия (разворот) гидроксила, занимающего положение sn-2.
В другом случае происходят три реакции. Предварительно sn-глицерол-З-фосфат окисляется в диоксиацетон-3-фосфат (который может быть получен и путем восстановления глицеральдегид-3-фосфата). Диоксиацетон-3-фосфат образует с GGDP простой эфир — 1-моноалкенил-диоксиацетонфосфат. Путем восстановления этого эфира образуется 3-моноалкенил-sn-глицерол-1-фосфат. В ходе реакции происходит инверсия гидроксила, занимающего положение sn-2.
Метаногенные и экстремально термофильные архей (рис. 157, Б). На примере Methanobacterium sp. и Sulfolobus sp. показано, что в результате альтернативной АТФ-зависимой киназной реакции sn-глицерол превращается в sn-глицерол-1-фосфат. Затем sn-глицерол-1-фосфат взаимодействует с GGDP, в результате чего образуется 3-моноалкенил-sn-глицерол-1-фосфат.
После добавления к 3-моноалкенил-sn-глицерол-1-фосфату второй молекулы GGDP из него образуется 2,3-диалкенил-sn-глицерол-1-фосфат, или преархетидная кислота (англ. pre-archaetidic acid).
Присоединение полярных головок к преархетидной кислоте. На данном этапе в положение 1 вводятся заряженные или электронейтральные полярные головки. Они присоединяются к глицеролу напрямую или через фосфорильный остаток. К их числу относятся глицерол, глицеросульфат, глицерофосфат, серин и этанол- амин, а также полиолы и простые сахара (см. I том учебника).
Образование 2,3-дифитанил-диглицерида (археола). На этапе образования археола из преархеола происходит множественное восстановление двойных связей. В результате этого геранил-геранильные радикалы превращаются в фитаниль- ные. Необходимо подчеркнуть, что гидрирование двойных связей редко встречается в биологических системах. Обычно происходит противоположно направленный процесс — увеличение количества двойных связей (десатурация).
Образование 2,3,2',3'-дибифитанил-тетраглицерида (кальдархеола). Простейший тетраэфир, или 2,3,2',3'-дибифитанил-тетраглицерид синтезируется за счет образования ковалентной связи между фитанильными хвостами двух молекул 2,3-дифитанил-диглицерида.
Способ, которым сшиваются друг с другом две Сrо-полиизопреновые цепи, не знает аналогий в биохимии полиизопреноидов, поскольку при образовании С40- предшественника каротиноидов — фитоина — две молекулы GGDP соединяются способом «голова к голове». Здесь же два фитанила соединяются способом «хвост к хвосту».
Образование циклопентановых колец внутри полиизопреноидной цепи. Механизм, посредством которого в бифитанильные цепи вводятся циклопен-
тановые кольца, неизвестен. Однако регулярность расположения указывает на то, что они образуются в аксиальносимметричном тетраэфире, а не на стадии свободных С20- или С40-хвостов, еще не присоединенных к глицеролу. Напоминаем, что максимальное число таких колец достигает восьми (см. I том учебника).
Биосинтез девятиатомного полиола (кальдитола). Девятиатомный кальдитол образуется путем слияния триозы (глицеральдегида или диоксиацетона) и гексозы (глюкозы или фруктозы) с последующим восстановлением кетогрупы в спиртовую.
15.2. Биосинтез полиизопреноидов
Полиизопреноиды представляют собой гидрофобные соединения, которые в большинстве случаев содержат систему сопряженных двойных связей. Благодаря особенностям своей молекулярной структуры они выполняют ряд важнейших биологических функций:
— формируют унитарные биомембраны (только у архей) или дополнительно стабилизируют их (у всех живых организмов);
— универсально предохраняют субклеточные структуры от окислительного стресса;
— используются в качестве вспомогательных пигментов, входя в состав светособирающих антенных комплексов;
— образуют гидрофобный домен, или хвост молекулы (бактерио)хлорофилла;
— обеспечивают индивидуальную окраску, выступая в роли одного из факторов поведенческой адаптации у высших организмов, а также способствуют расселению низших организмов (примером служат плодовые тела миксобактерий).
Полиизопреноиды можно формально рассматривать в качестве продуктов полимеризации изопрена (метилбутадиена) с появлением двойной связи на центральной оси мономера:
n х [СН2=С(СН3)-СН=СН2] —> [-СН2-С(СН3)=СН-СН2-]n.
При сборке полиизопреновой цепи в качестве мономера используется, вопреки названию, не изопреновая группа, а ее активированный предшественник — изопен- тенилдифосфат (англ. isopentenyl diphosphate, IDР):
СН2=С (СН3)-СН2 - СН2 - ОРР.
По завершении сборки полиизопреновая цепь подвергается дополнительным превращениям, важнейшие из которых — изменение степени насыщенности и циклизация.
Изменение степени насыщенности. Полиизопреновая цепь либо десатурируется (т. е. в ней появляются дополнительные двойные связи), либо сатурируется (т. е. двойные связи превращаются в одинарные).
В первом случае одинарная связь между соседними изопреновыми группами превращается в двойную:
[-СН2-С(СН3) =СН-СН2-СН2-С(СН3) =СН-СН2-] —> [-СН2-С(СН3) =СН-СН=СН-С(СН3) =СН-СН2-] + 2Н+/2е-.
Такую стратегию процессинга полиизопреноидов в той или иной степени используют все живые организмы, когда они синтезируют каротиноиды и их производные.
Во втором случае двойные связи в соседних изопреновых группах превращаются в одинарные:
[CH2-С(СН3) =СН-СН2-СН2-С(СН3) =СН-СН2-] + 4Н+/4е- —> [-СН2-СН(СН3)-СН2-СН2-СН2-СН(СН3)-СН2-СН2-].
Такой стратегией процессинга полиизопреноидов пользуются только археи при образовании гидрофобных хвостов своих уникальных мембранных липидов.
Циклизация. В полиизопреноидной цепи могут содержаться отдельно расположенные внутренние кольца (в кальдархеолах архей; см. I том учебника), а также терминальные циклопентановые кольца (в к-каротиноидах), циклогексановые кольца (в β- и ε-каротиноидах) или арильные кольца (в![]() каротиноидах).
каротиноидах).
Альтернативный вариант циклизации — это образование сотовидных тетрациклических или пентациклических структур, которые называются соответственно стероидами и гопаноидами (см. I том учебника).
Введение заместителей. В качестве боковых заместителей в полиизопреноиды могут вводиться различные кислородсодержащие группы (гидрокси-, кето-, эпокси-, метокси-, формил-, карбокси- и гликозидная).
Классификация полиизопреноидов. Полиизопреноиды можно подразделить на каротиноиды и не-каротиноиды.
Каротиноиды состоят из восьми или большего числа изопреновых единиц. В их основе лежит центрально-симметричная структура. Согласно классическому определению, данному Паулем Каррером, каротиноидами называются такие полиизопреноиды, у которых две центральные метильные группы разделены шестью атомами углерода, а прочие соседствующие метильные группы — пятью атомами углерода (см. рис. 133).
Не-каротиноиды имеют в своей основе асимметричную структуру. Они могут быть ациклическими, могут содержать внутри цепи, изолированные циклопентановые кольца (оба случая связаны с гидрофобными хвостами полярных липидов архей; см. I том учебника), могут содержать терминальное арильное кольцо (хиноны; см. рис. 77), а также могут состоять из трех-четырех конденсированных циклогексановых колец, сросшихся с одним циклопентановым кольцом (гопаноиды и стероиды).
Каротиноиды. Каротиноиды в основном гидрофобны. Поэтому они чаще всего входят в состав гидрофобного ядра унитарной мембраны или нековалентно связываются с белками. У (ан)оксигенных фототрофных бактерий они образуют тройственные (бактерио)хлорофилл-каротиноид-белковые комплексы. У хемотрофных бактерий функции каротиноидов менее специфичны. Они, прежде всего, связаны со структурной стабилизацией мембран, а также их защитой от окислительного повреждения.
В разделе 12.6, посвященном фотосинтетическим пигментам, мы уже кратко охарактеризовали каротиноиды. Теперь нам предстоит более подробно рассмотреть их свойства, а также остановиться на путях их биосинтеза.
Каротиноиды — это наиболее распространенные природные пигменты, которые придают индивидуальную окраску или расцветку большинству прокариотных организмов, протистов, растений, грибов и животных.
Цвет каротиноидов зависит от числа сопряженных двойных связей, по мере увеличения которого максимум поглощения сдвигается в длинноволновую часть видимого спектра. Количество сопряженных двойных связей увеличивается на ранних этапах биосинтеза каротиноидов, причем чем «старше» каротиноид, тем сильнее он окрашен и тем «краснее». Поздние этапы биосинтеза окрашенных каротиноидов связаны с их «доработкой» путем образования циклических групп на одном или обоих концах молекулы, а также за счет добавления к полиизопреновому скелету кислородсодержащих заместителей (см. выше). Результатом является обогащение спектральных свойств и исключительное разнообразие структуры природных каротиноидов (>700 молекулярных форм).
Окрашенные каротиноиды служат незаменимыми компонентами фотосинтетического аппарата как бактериородопсинового, так и хлорофильного типа (см. главу 12). При этом они выполняют три основные функции.
Функция вспомогательного светособирающего, или антенного пигмента. На свету каротиноиды поглощают кванты в сине-зеленой части спектра (400-550 нм), переходят в возбужденное состояние и путем электромагнитного резонанса возбуждают молекулы (бактерио) хлорофилла, удаленные от них на расстояние <5А.
Структурная функция. Каротиноиды участвуют в процессе самосборки фотосинтетических пигмент-белковых комплексов, а затем поддерживают их в нативном состоянии.
Защитная функция. Мы уже говорили о том (см. раздел 12.6.1), что при избыточном накоплении возбужденных синглетных молекул хлорофилла они переходят в долгоживущее триплетное состояние и начинают физически взаимодействовать с молекулярным кислородом. При этом неактивный триплетный кислород (3О2) переходит в активное синглетное состояние (3О2), что становится причиной окислительного стресса и фотоповреждения. Каротиноиды способны перехватывать возбужденные синглетные и триплетные состояния хлорофилла, а также возбужденное синглетное состояние кислорода. В результате они сами переходят в возбужденное синглетное или триплетное состояние, а затем избыточная энергия освобождается в виде квантов флуоресценции или рассеивается в форме тепла (подробнее см. III том учебника). Для того, чтобы эти процессы стали достаточно эффективными, в молекуле каротиноида должно быть не менее семи сопряженных двойных связей, т. е. он должен быть окрашен.
15.2.1. Биосинтез мономера полиизопреноидов — изопентенилдифосфата
Известны два разных пути биосинтеза IDP (рис. 158). Первый, так называемый «мевалонатный» путь был открыт в 1960-е годы Феодором Линеном при изучении биосинтеза холестерола. В настоящее время показано, что такой путь реализуется у архей, а также в цитозоле животных и высших растений.
Рис. 158. Пути биосинтеза IDP, или изопреновой группы (по: Коуаша, 2002). А — мевалонатный путь. HMG-CoA — 3-окси-3-метил-глутарил-СоА; IDP — изопентенилдифосфат; 1—ацетил-СоА-ацетилтрансфераза; 2 — HMG-CoA-синтаза; 3 — HMG-CoA-редуктаза; 4 — мевалонат- киназа; 5 — фосфомевалонат-киназа; 6 — мевалонилдифосфат-декарбоксилаза. Б — альтернативный путь. DXP — 1-дезокси-D-ксилулозо-5-фосфат; ЦТФ, ЦДФ и ЦМФ — цитидинтрифосфат, цитидинди- фосфат и цитидинмонофосфат; МЕР — 2-метил-D-эритритол-4-фосфат; МЕ — 2-метил-D-эритритол; MECDP — 2-метил-D-эритритол-2,4-циклодифосфат; HMBDP — 1-окси-2-метил-2-бутенил-4-дифосфат; 1 — DXP-синтаза; 2 — DXP-редуктоизомераза; 3 — МЕР-цитидилтрансфераза; 4 — ЦДФ-МЕ-киназа; 5 — MECDP-синтаза; 6 — HMBDP-синтаза.
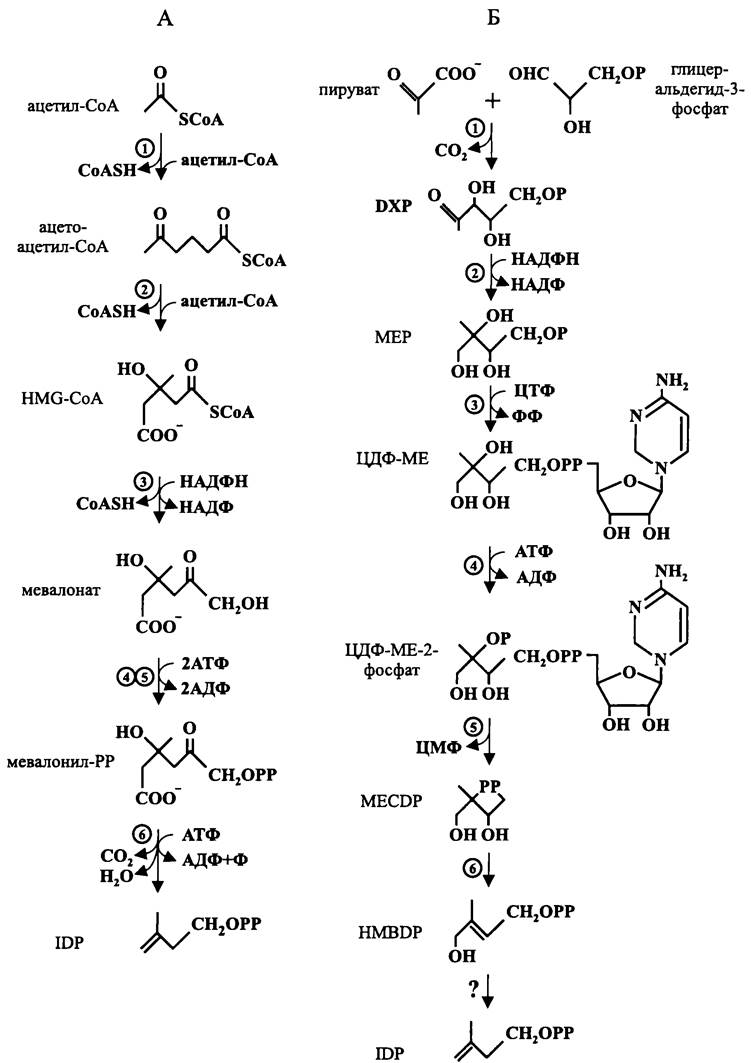
Мевалонатый путь (рис. 158, А). В данном случае исходным субстратом служит ацетил-СоА.
Из двух молекул ацетил-СоА с помощью ацетил-СоА-ацетилтрансферазы образуется ацетоацетил-СоА. После присоединения к нему третьей молекулы ацетил-СоА он превращается в 3-окси-3-метил-глутарил-СоА (англ. 3-hydroxy-3-methyl- glutaryl-CoA, HMG-CoA). Затем НАДФН-зависимая HMG-CoA-редуктаза восстанавливает З-гидрокси-З-метил-глутарил-СоА в 3,5-диоксо-3-метил-пентановую, или мевалоновую кислоту (англ. mevalonic acid). С помощью мевалонаткиназы, а затем фосфомевалонат-киназы мевалоновая кислота превращается в мевалонилдифосфат. В конечном счете, из мевалонилдифосфата под воздействием АТФ-зависимой мевалонилдифосфат-декарбоксилазы образуется IDP.
После открытия мевалонатного пути решили, что он универсален для всех живых организмов. Однако в начале 1990-х годов выяснилось, что бактерии (в частности, Е. coli) не включают меченый ацетат в полиизопреноиды. Это указывало на существование альтернативного пути биосинтеза IDP.
Альтернативный путь (рис. 158, Б). Первую реакцию альтернативного «не- мевалонатного» пути обнаружил в 1996 г. у Е. coli немецкий биохимик Ромер (M. Rohmer). Остальные изучил японский молекулярный биолог Кояма (T. Коуата). В настоящее время показано, что этот путь используют многие бактерии, а также зеленые водоросли, хлоропласты высших растений и некоторые протисты (в частности, Plasmodium sp.).
В данном случае исходными субстратами служат неактивированный пируват и глицеральдегид-3-фосфат, которые конденсируются с образованием 1-дезокси-D- ксилулозо-5-фосфата (англ. 1-deoxy-D-xylulose-5-phosphate, DXP). Реакция включает в себя декарбоксилировение, и ее катализирует DXP-синтаза, которая использует в качестве кофактора тиамин (см. рис. 89), а также катионы Mg2+и Мn2+.
На втором этапе сопряженно происходят восстановление и изомеризация DXP с образованием 2-метил-D-эритритол-4-фосфата (англ. 2-methyl-D-erythritol- 4-phosphate, МЕР). Эту реакцию катализирует DXP-редуктоизомераза.
DXP-редуктоизомераза широко распространена у многих бактерий, в том числе патогенов, а также у растений, однако она отсутствует у животных, в частности млекопитающих. Специфическим ингибитором этого фермента служит антибиотик фозмидомицин. Он и антибиотики с аналогичным механизмом действия являются сильнодействующими агентами против грамположительных и грамотрицательных бактерий, а также перспективными гербицидами.
Третья реакция альтернативного пути связана с переносом МЕР на цитидин- трифосфат с образованием (цитидин-5'-дифосфо)-2-метил-D-эритритола, или ЦДФ- МЕ. Эту реакцию катализирует МЕР-цитидинтрифосфат-трансфераза. На четвертом этапе ЦДФ-МЕ-киназа фосфорилирует МЕ в положении 2. После этого от ЦЦФ- МЕ-2-фосфата отщепляется ЦМФ. При этом из МЕ образуется цикло-фосфодиэфир (англ. 2-methyl-D-erythritol-2,4-cyclodiphosphate, MECDP). Катализатором реакции служит MECDP-синтаза. На шестом этапе MECDP превращается в ациклический дифосфат — 1-окси-2-метил-2-бутенил-4-дифосфат (англ. l-hydroxy-2-methyl-2- butenyl-4-diphosphate, HMBDP). На последнем этапе HMBDP превращается в IDP. Эту реакцию катализирует еще не идентифицированный фермент, который кодируется геном lytB.
15.2.2. Магистральный путь биосинтеза полиизопреноидов и его ответвления
Магистральный путь биосинтеза полиизопреноидов универсален для представителей доменов Bacteria, Archaea и Еuсаrуа. Это дает основание считать, что они унаследовали его от гипотетического общего предка-прогенота (см. I том учебника).
Ферменты, участвующие в магистральном пути биосинтеза полиизопреноидов (рис. 159), высококонсервативны. Их гены у прокариотов расположены на хромосоме. У ядерных организмов они находятся в ядре, а у растений — частью в ядре, частью в пластидах.
Первый шаг в расшифровке пути биосинтеза каротиноидов и объяснении их биологической роли совершил французский микробиолог Стэниер (R. Y. Stanieг). При изучении в 1955 г. «сине- зеленого» мутанта Rhodobacter sphaeroides R-26 он установил, что окрашенные каротиноиды образуются в результате десатурации бесцветных предшественников и оказывают защитное действие против окислительного фотоповреждения.
Следующим крупным шагом были исследования американского генетика Мерса (В.Маrrs), который в начале 1980-х годов разработал системы для изучения генетической рекомбинации у пурпурных бактерий из класса «Alphaproteobacteria», а также работы японского генетика Мисавы (N.Misawa), который в начале 1980-х годов реконструировал пути биосинтеза каротиноидов у фитопатогенной бактерии Erwinia herbicola и изучил экспрессию гетерологичных генов каротиногенеза в клетках Е. coli.
Хотя мономером при сборке полиизопреновой цепи служит IDP, в качестве затравки-праймера используется не он сам, а продукт его изомеризации — диметилаллил-дифосфат (англ. dimethylallyl diphosphate, DMADP):
СН2=С(СН3)-СН2-СН2-ОРР —> (СН3)2С=СН-СН2-ОРР.
Диметилаллил-трансфераза переносит IDР на DMADP с образованием геранил- дифосфата (англ. geranyl diphosphate, GDP). При этом двойная связь в IDР перемещается из терминального положения в среднее:
СН2=С(СН3)-СН2-СН2- —> -СН2-С(СН3) = СН-СН2-. Таким образом, GDP состоит из двух изопреновых групп:
СН3-С(СН3) = СН-СН2- и -СН2-С(СН3) = СН-СН2-.
GDP и эквивалентный ему десятиуглеродный блок, входящий в состав диизопреноидов, или дипреноидов называются монотерпенами (от греч. terebinthos — скипидарное дерево). Пятнадцатиуглеродные трипреноиды называются соответственно сесквитерпенами (от лат. sesqui — в полтора раза), двадцатиуглеродные тетрапреноиды—дитерпенами, двадцатипятиуглеродные пента- преноиды — сестерпенами, тридцатиуглеродные гексапреноиды — тритерпенами, тридцатипятиуглеродные гептапреноиды — дитерсестерпенами, сорокауглеродные октапреноиды—тетратерпенами и т. д., а в целом для обозначения полиизопреноидов есть синоним — терпеноиды.
Дальнейшие этапы биосинтеза полиизопреноидов заключаются, в первую очередь, в наращивании цепи новыми мономерами ГОР при помощи элонгирующих ферментов — пренилтрансфераз, или синтаз. В результате могут образовываться как cis-, так и trans-изомеры.
В настоящий момент охарактеризовано до шестнадцати пренилтрансфераз, относящихся к четырем основным группам. Эти ферменты образуют полиизопреновые цепи с разной конфигурацией и разным количеством атомов углерода — от n=10 (гераниол) до n>5 • 103 (натуральный каучук).
Пренилтрансферазы I синтезируют írnns-полиизопреноиды с относительно короткой цепью (от С15 до С20).
Пренилтрансферазы II образуют frans-цепи средней длины (от С25 до С35).
Пренилтрансферазы III обеспечивают образование длинноцепочечных trans-продуктов (С40 и выше).
С помощью пренилтрансфераз IV типа образуются длинноцепочечные мульти- сis-продукты. Для действия элонгирующих ферментов всех четырех типов требуются катионы Mg2+и Мn2+.
Пренилтрансфераза I (синтаза I) отвечает за удлинение цепи в такой последовательности: С10 (GDP) —> С15 (фарнезилдифосфат; англ. farnesyl diphosphate, FDP) —> С20(геранилгеранил-дифосфат; англ. geranylgeranyl diphosphate, GGDP). Присоединение каждого нового остатка IDР происходит способом «хвост к голове» (рис. 159).
Затем эта строгая последовательность прерывается, и фитоинсинтаза CrtB соединяет две молекулы GGDP по способу «голова к голове». В результате образуется первый тетратерпен — бесцветный С40-каротиноид 15,15'-сis-фитоин, который содержит только три сопряженные двойные связи (принцип нумерации атомов углерода в молекуле полиизопреноидов указан в разделе 12.6.2 и изображен на рис. 133). Следует отметить, что большинство природных каротиноидов — это полные trans-изомеры. Важнейшим исключением является фитоин. Он обычно существует в форме 15,15′-cis-изомера.
Рис. 159. Магистральный путь биосинтеза полиизопреноидов. IDP — изопентенилдифосфат; DMADP — диметилаллил-дифосфат; GDP — геранил- дифосфат; FDP — фарнезилдифосфат; GGDP — геранилгеранил-дифосфат; PPDP — префитоин-дифосфат; 1 — IDР-изомераза; 2 — DMADP-трансфераза; 3 — FDP-синтаза; 4 — GGDP-синтаза; 5 — фитоинсинтаза.

Предшественником фитоина служит нестабильный интермедиат — префитоин-дифосфат (англ. prephytoene diphosphate, PPDP). Его образование представляет собой первую реакцию, специфичную для пути биосинтеза С40-каротиноидов.
Путем дальнейших молекулярных преобразований из фитоина образуются С40-, С45- и С50-каротиноиды (см. ниже).
Первая боковая ветвь (от FDP). Продуктами этой ветви становятся исключительно важные в биологическом отношении сесквитерпены, тритерпены и высшие терпеноиды.
Прежде всего, из FDP образуются два C15-спирта. Первый из них — это ненасыщенный спирт фарнезол. Он выполняет роль гидрофобного хвоста в молекуле бактериохлорофиллов с-е (см. раздел 12.6.1). Второй спирт — это насыщенный фар- незанол. Он выполняет роль гидрофобного хвоста в одном из вариантов полярных липидов у экстремально галофильных архей. Наконец, из FDP образуется насыщенный углеводород фарнезан, который относится к числу неполярных липидов у архей (см. I том учебника).
Кроме того, на боковой ветви начинается так называемый С30-путь. Синтаза CrtM соединяет две молекулы FDP способом «голова к голове», в результате чего образуется первый тритерпен —дегидросквален, или диапофитоин («фитоин, укороченный с обоих концов»). Его производными являются:
— С30-каротиноиды, в частности сквален; он редко встречается у бактерий (примерами служат Heliobacterium sp. и Staphylococcus sp.), однако у архей это характерный мембранный липид;
— хиноны; они входят в состав цитохром bс1(b6f)-комплекса и содержат от семи до десяти изопреновых групп (см. раздел 10.4.6 и рис. 77);
— полиеновые спирты ундекапренол и долихол; первый из них содержит одиннадцать изопреновых групп (С55) и у прокариотов служит переносчиком субъединиц муреина, псевдомуреина и капсульных полисахаридов (см. I том учебника); второй содержит от четырнадцати до двадцати двух изопреновых групп (С70—С110) и у эукариотов служит переносчиком олигосахаридов, которые используются при биосинтезе поверхностных гликопротеинов.
Из сквалена синтезируются неполярные липиды разной степени восстановленности, вплоть до предельного углеводорода сквалана (характерен для архей).
Кроме того, сквален служит предшественником для биосинтеза двух классов циклических терпеноидных липидов — тетрациклических стероидов (типичны для эукариотов, однако редко встречаются у прокариотов) и пентациклических гопаноидов (широко распространены у бактерий, но не встречаются у архей и эукариотов).
Вторая боковая ветвь (от GGDP). Хотя продукты этой ветви не столь многообразны, как у первой, их биологическое значение также велико.
GGDP служит предшественником при образовании ненасыщенных С20-спиртов, которые содержат соответственно четыре, две и одну двойную связь. Первый спирт — это геранилгераниол, который выполняет роль гидрофобного хвоста бактериохлорофилла g. Второй спирт — это фитодиенол, он образует гидрофобный хвост бактериохлорофилла b.Третий спирт — это фитол, входящий в качестве гидрофобного хвоста в состав бактериохлорофиллов а и b, а также хлорофиллов а, a2, b, b2 и d (см. раздел 12.6.1).
Путем полной сатурации из GGDP синтезируется спирт фитанол. Он служит боковым радикалом в полярных липидах архей (см. I том учебника).
Биосинтез ациклических и циклических каротиноидов. Большинство природных каротиноидов — это С4о-каротиноиды. Редки и мало изучены С35- каротиноиды, которые образуются путем конденсации FDP и GGDP, а также гомокаротиноиды (С>40).
Как уже указывалось, путь биосинтеза С40-каротиноидов начинается с 15,15′- cis-фитоина, содержащего три сопряженные двойные связи (рис. 160). Десатурация фитоина сопровождается спонтанной изомеризацией с образованием полных trans- продуктов.
Рис. 160. Путь биосинтеза С40-каротиноидов. Фитоин для удобства изображен в 15-15'-trans конфигурации. 1—десатураза СrtI; 2 — гидратаза СrtС; 3 — десатураза СrtD; 4 — метилаза СгtF; 5 — циклаза СrtY.

Вначале бесцветный фитоин трижды десатурируется и через стадию желтоватого![]() каротина (семь сопряженных двойных связей) превращается в бледно-желтый нейроспорин (девять сопряженных двойных связей). Катализатором этой реакции служат фитоиндесатуразы.
каротина (семь сопряженных двойных связей) превращается в бледно-желтый нейроспорин (девять сопряженных двойных связей). Катализатором этой реакции служат фитоиндесатуразы.
В ходе эволюции появились два негомологичных типа фитоиндесатураз. У большинства бактерий, архей и грибов имеются НАДФ- или ФАД-зависимые фитоиндесатуразы Crtl. Отдельный кластер образуют пластохинон-зависимые фитоиндесатуразы — фитоиндесатураза CrtP цианобактерий и фитоиндесатураза Pds водорослей и высших растений.
Фитоиндесатуразы Crtl вводят в фитоин три двойные связи (с образованием нейроспорина в случае Rhodobacter sphaeroides) или четыре двойные связи (с образованием ликопина в случае Erwinia herbicola и Rhodospirillum rubrum).
Фитоиндесатуразы CrtP и Pds вводят только две двойные связи (с образованием![]() -каротина). Для превращения
-каротина). Для превращения![]() каротина в нейроспорин, а нейроспорина в ликопин, т. е. для введения соответственно третьей и четвертой двойной связи, требуются особые
каротина в нейроспорин, а нейроспорина в ликопин, т. е. для введения соответственно третьей и четвертой двойной связи, требуются особые![]() каротиндесатуразы I
каротиндесатуразы I![]() каротиндесатураза CrtQb цианобактерий и растительная
каротиндесатураза CrtQb цианобактерий и растительная![]() -каротиндесатураза Zds).
-каротиндесатураза Zds).
На стадии нейроспорина магистральный путь биосинтеза каротиноидов разветвляется (рис. 160).
Ликопиновая ветвь. У одних бактерий, например, Е. herbicola, R. acidophila и R. rubrum, а также у растений нейроспорин еще раз десатурируется. В результате образуется розовый ликопин, содержащий одиннадцать сопряженных двойных связей.
Ациклические производные ликопина характерны для пурпурных бактерий. Они синтезируются с помощью гидратазы CrtC, десатуразы CrtD и мети лазы CrtF. Эти ферменты образуют двойные связи в положениях 3:4 и 3':4', а также вводят четвертичные гидроксильные и оксиметильные группы в положения 1 и 1'. Интермедиатами пути, который заканчивается образованием ярко-красного спириллоксан- тина (тринадцать сопряженных двойных связей), являются минорные каротиноиды родопин, 3,4-дидегидрородопин, ангидрородовибрин, родовибрин и монодеметил- спирилло ксантин.
Циклические производные ликопина, или каротины содержат β-иононовые или ε- иононовые концевые группы. Самым распространенным каротиноидом у Bacteria, Archaea и Еuсаrуаявляется β-каротин, содержащий две концевые β-иононовые группы. В результате β-циклизации на одном конце молекулы ликопин превращается в y- каротин. Его дополнительная β-циклизация приводит к образованию β-каротина (рис. 160).
Существуют, по крайней мере, три β-ликопинциклазы. Одна — это «классическая» гомодимерная СrtY-циклаза, которая имеется у хемотрофных бактерий. Другая — это гомодимерная СrtL- циклаза цианобактерий и растений. Третья — это гетерод и мерная СrtYсd-циклаза, обнаруженная у некоторых актинобактерий и археота Sulfolobusacidocaldarгus.
Альтернативная (гидроксинейроспориновая) ветвь. У других бактерий, например, R. sphaeroides, нейроспорин с помощью гидратазы СrtС превращается в гидрок- синейроспорин. Затем, как и при синтезе спириллоксантина в ликопиновой ветви, десатураза СrtD образует двойную связь в положении 3:4, а метилаза СrtF вводит четвертичную метальную группу в положение 1. По контрасту с симметричным спириллоксантином, конечным продуктом является асимметричный сфероидин. Этот красный каротиноид содержит одиннадцать сопряженных двойных связей. Интермедиатом на пути его образования является минорный каротиноид деметилсфероидин.
Оранжевый, коричневый и красный цвет, которым обладают популяции пурпурных бактерий, находит два объяснения. Во-первых, у них относительно много ярко окрашенных каротиноидов (до 60% содержания бактериохлорофилла). Во-вторых, in situ главные максимумы бактериохлорофиллов а или b лежат в инфракрасной части спектра (соответственно около 800 и 900 нм), и поэтому эти пигменты имеют бледно-голубой цвет. Для сравнения отметим, что для бактериохлорофиллов с, d и q, а также для всех хлорофиллов характерны более коротковолновые главные максимумы (порядка 700 нм), и эти пигменты по цвету сине-зеленые. Поэтому в случае низкого содержания вспомогательных пигментов, в частности каротиноидов, популяции зеленых бактерий, гелиобактерий и цианобактерий приобретают зеленую окраску.
Биосинтез ксантофиллов. Ксантофиллы, или кислородсодержащие каротиноиды образуются в результате появления в молекуле каротина одного или нескольких атомов кислорода в составе гидроксильной группы, кетогруппы, формильной группы или оксиметильной группы.
У пурпурных бактерий ксантофиллы синтезируются в анаэробных условиях путем гидратации двойных связей в положениях 1:2 и 1':2' (см. выше) или внутри цепи; кетогруппы появляются в результате окисления гидроксильных групп. Формильные и метоксильные группы образуются при помощи монооксигеназ только в аэробных условиях.
У цианобактерий после гидроксилирования в положениях 3 и 3' α-каротина или β-каротина образуются характерные ксантофиллы β-криптоксантин и зеаксантин. Для действия гидроксилаз необходим молекулярный кислород и катионы Fе2+. Ке- токаротиноиды пурпурных бактерий синтезируются при помощи монооксигеназ, которые вводят атом кислорода в положения 2 или 4 (см. рис. 133).
К числу ксантофиллов формально можно отнести и гликозидные каротиноиды (см. рис. 133). Они синтезируются при помощи соответствующих гликозилаз — например, зеаксантингликозилаза превращает зеаксантин в моно- или дигликозиды.
15.3. Биосинтез тетрапирролов
Тетрапирролы содержат более или менее протяженную систему двойных связей (см. рис. 120) и поглощают свет в видимой части спектра. Многие из них образуют порфириновый макроцикл, способный лигандировать катионы металлов или небольшие органические молекулы. Благодаря этим особенностям молекулярной структуры тетрапирролы могут выполнять разнообразные и исключительно важные биологические функции:
— поглощать видимый свет при фототрофии и фотоповедении, т. е. выступать в роли фоторецептора (бактериохлорофиллы, хлорофиллы и фикобилины фотосинтетических светособирающих антенн, а также фитохромы);
— сопрягать фотофизические процессы с фотохимическими, т. е. выступать в роли фотосенсибилизаторов (хлорофилл или бактериохлорофилл в качестве первичного донора фотосинтетического реакционного центра);
— векторно переносить электроны, т. е. выступать в роли компонентов электрон- транспортных цепей (бактериохлорофиллы и хлорофиллы фотосинтетических реакционных центров, а также гемы цитохромов);
— переносить метильную группу, т. е. выступать в роли кофактора трансметилаз (коррин, F430).
Тетрапирролы синтезируются с помощью ферментативных реакций, которые принадлежат к одному и тому же разветвленному метаболическому пути.
Главными модельными объектами при изучении биосинтеза тетрапирролов и его общей регуляции послужили представители класса «Аlphaproteobacteria». Сравни-
тельные исследования, проведенные на цианобактериях, зеленых водорослях и высших растениях, показали, что важнейшие этапы этого пути и катализирующие их ферменты стандартны для всех живых организмов. У прокариотов гены ферментов, с помощью которых образуются конечные продукты пути биосинтеза тетрапирро- лов, расположены на хромосоме. У ядерных организмов они находятся в ядре, а у растений — частью в ядре, частью в пластидах.
Биосинтез ALA — предшественника тетрапирролов. Универсальным предшественником тетрапирролов служит 5-аминолевулиновая, или δ-аминолевулиновая кислота (англ. δ-aminolevulinic acid, ALA). Пурпурные бактерии из класса «Alphaproteobacteria», некоторые бактерии из филы ВХIII Firmicutes, а также грибы синтезируют ALA путем конденсации глицина с сукцинил-СоА. В то же время большинство бактерий, а также археи, водоросли и высшие растения образуют ALA из глутаминовой кислоты (рис. 161, А). Особенностью этого пути является участие в нем tPHKglu, которая присоединяется к глутаминовой кислоте и тем самым ее активирует. Катализатором реакции служит Lглу-тРНКGlu-синтаза. Следует подчеркнуть, что РНК крайне редко участвует в энзиматических процессах, не приводящих к образованию пептидной связи.
С помощью НАДФН-зависимой Lглу-тРНКСlu-редуктазы активированный глутамат восстанавливается до полуальдегида (англ. glutamate semialdehyde, GSA). Эта реакция происходит только в присутствии катионов Mg2+. На заключительном этапе аминогруппа GSA переносится из положения 4 в положение 5, в результате чего образуется ALA. Кофактором этой аминотрансферазной реакции является пиридоксальфосфат (рис. 162), который представляет собой альдегидное производное спирта пиридоксола, или витамина В6. Между альдегидной группой пиридоксальфосфата и переносимой им аминогруппой временно образуется шиффово основание (см. раздел 10.4.1, а также рис. 73). Возможно, что все три фермента, участвующие в синтезе ALA, входят в один мультиферментный комплекс.
Биосинтез протопорфирина IX. Вначале восемь молекул ALA попарно конденсируются друг с другом с образованием четырех пиррольных колец порфобилиногена (рис. 161, Б). Катализатором этой реакции служит Mg2+- зависимая ALA-дегидратаза. Затем четыре молекулы порфобилиногена одновременно дезаминируются, и из них получается нестабильный ациклический тетрапиррол — гидроксиметилбилан. Данную реакцию катализирует порфобилиноген- дезаминаза, а ее кофактором является димер порфобилиногена. Следом за этим гидроксиметилбилан при помощи уропорфириноген III-синтазы, или «косинтазы» превращается в макроциклический тетрапиррол — уропорфириноген III. Дезаминаза и косинтаза, скорее всего, образуют бинарный функциональный комплекс.
Рис. 161. Пути биосинтеза 5-аминолевулиновой кислоты (А) и протопорфирина IX (Б). А: 1 — Lглу-тРНКGlu-синтаза; 2 — Lглу-тРНКGlu-редуктаза; 3 — GSА-аминотрансфераза. Б: 1 — АLА- дегидратаза; 2 — порфобилиноген-дезаминаза; 3 — уропорфириноген III-синтаза (косинтаза); 4 — уропор- фириноген III-декарбоксилаза; 5 — оксидаза/декарбоксилаза; 6 — протопорфириноген IХ-оксидаза.
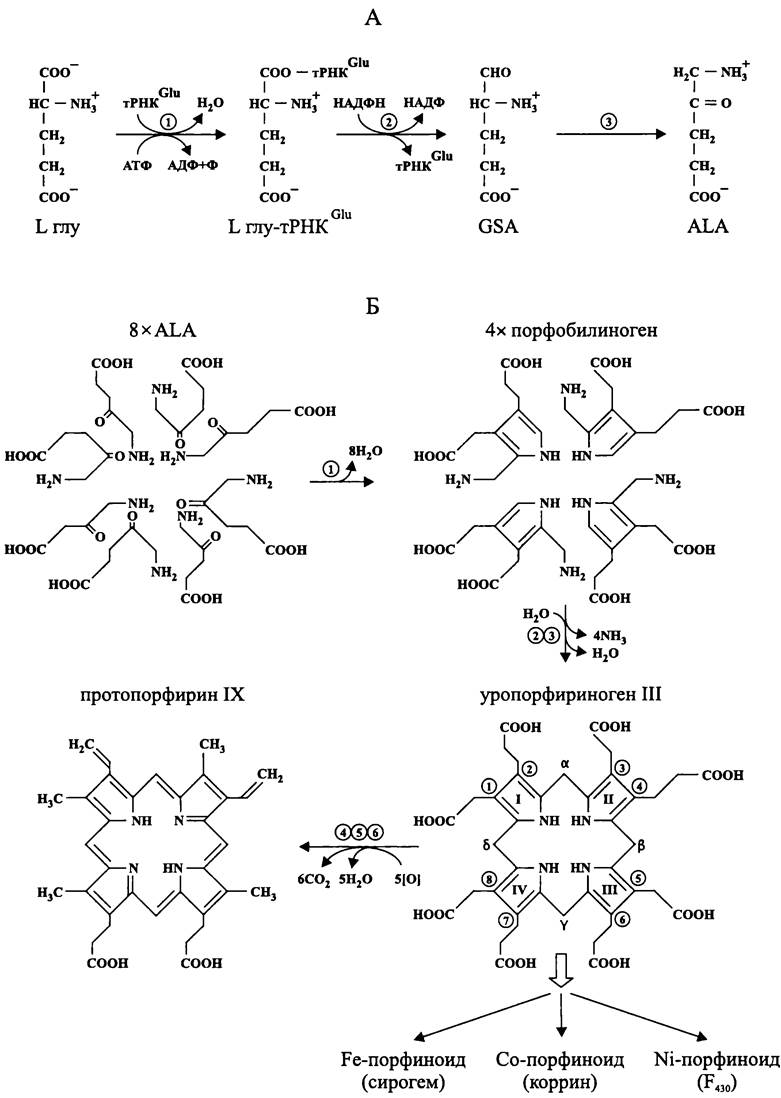
Уропорфириноген III служит предшественником для трех исключительно важных металпорфиноидов.
Во-первых, это Fe-порфиноид — сирогем (один из двух вариантов гема, входящего в качестве простетической группы в состав цитохрома d; см. раздел 10.4.1, а также рис. 70).
Во-вторых, это Со-порфиноид — коррин (простетическая группа трансметилаз у метаногенных архей и ацетогенных бактерий; см. разделы 11.4.5.4 и 11.4.5.5, а также рис. 80).
Втретьих, это Ni-порфиноид — F430 (простетическая группа метил-СоМ редуктазы у метаногенных архей; см. раздел 11.4.5.4, а также рис. 119).
Реакции, приводящие к образованию этих производных уропорфириногена III, включают в себя: (1) метилирование порфиринового ядра в положениях 1 и 3; (2) окисление прекоррина II до тетрагидропорфирина; (3) восстановление макроцикла с максимальным уменьшением протяженности системы сопряженных двойных связей в случае F430 (см. рис. 120); (4) хелатирование катионов Fе2+, Со2+ и Ni2+ при помощи соответствующих хелатаз.
Из уропорфириногена III через две промежуточные стадии образуется протопорфирин IX.
Сначала под воздействием декарбоксилазы, которая удаляет карбоксильные группы в положениях 1, 3, 5 и 8, уропорфириноген III превращается в копропорфириноген III. Декарбоксилирование начинается с пиррольного кольца IV и продолжается по часовой стрелке, т. е. последовательно в положениях 8 —> 1 —> 3 —> 5.
Следом за этим оксидаза/декарбоксилаза окислительно декарбоксилирует пропионильные группы в положениях 2 и 4. Все они превращаются в винильные, в результате чего образуется протопорфириноген IX. Реакция протекает либо при участии молекулярного кислорода, либо в анаэробных условиях.
В завершение всего протопорфириноген IX-оксидаза образует метинильные мостики в положениях α, β, y, и δ, а также двойные связи в положениях 3:4 и 7:8. В результате этого образуется протопорфирин IX, который содержит систему сопряженных двойных связей и имеет красную окраску. Он служит предшественником при биосинтезе гемов, фикобилинов и (бактерио)хлорофиллов.
Биосинтез гемов. Ветвь железопорфиноидов, или гемов начинается (за исключением сирогема) с превращения протопорфирина IX в протогем.
Затем протогем-ферролиаза, или Fе2+-хелатаза катализирует образование комплекса между протопорфирином IX и катионом Fe2+. Кофакторами этой реакции служат катионы Со2+ и Zn2+. После этого из протогема образуются гемы а-d (см. рис. 70).
Биосинтез фикобилинов. Фикобилины представляют собой квази-циклические тетрапирролы в виде разрезанного кольца, которые не хелатируют катионы металлов (см. раздел 12.6.3). При связывании с апопротеином оно распрямляется, и образуется линейный тетрапиррол (см. рис. 134).
В образовании фикобилинов из протогема участвуют гемоксигеназы, редуктазы и изомеразы.
Первый этап на пути биосинтеза фикобилинов катализируется двухкомпонентной ферментной системой, состоящей из гемоксигеназы и НАДФ, или ферредоксинзависимой редуктазы. Гемокси- геназа размыкает порфириновый макроцикл протогема по метинильному мостику α между I и IV пиррольными кольцами, в результате чего образуется желчеподобный пигмент — биливердин IХα. Затем редуктаза I восстанавливает метинильный мостик между III и IV пиррольными кольцами, и биливердин IXα превращается в 15,16-дигидробиливердин IХα.
Рис. 162. Пиридоксальфосфат. Стрелкой указано место присоединения аминогруппы.

На втором этапе редуктаза II восстанавливает I пиррольное кольцо, и из 15,16- дигидробиливердина IХα образуется ЗZ-фикoэpитpoбилин. При помощи редуктазы III, которая восстанавливает винильную группу при IV пиррольном кольце в этильную, 3Z-фикоэритробилин превращается в 3Z-фикoциaнoбилин. 3Z-изомеры фикобилинов нестабильны, и поэтому фикобилин- этилиден-сis/trans-изомераза трансформирует их соответственно в 3Е-фикоэритробилин и 3Е- фикоцианобилин.
Для того, чтобы ковалентно присоединить фикобилин к апопротеину, используются специальные лигазы. Например, фикоцианобилин связывается через I пиррольное кольцо с аминокислотным остатком Суs-84, входящим в состав α-субъединицы (см. раздел 12.6.3, а также рис. 134).
Биосинтез (бактерио)хлорофиллов. Ветвь магнийпорфиноидов, или хлорофиллов начинается с превращения протопорфирина IX в 2,4-дивинил- протохлорофиллид (рис. 163). Образование комплекса между протопорфирином IX и катионом Mg2+ катализирует АТФ-зависимая Mg2+-хелатаза. Катион Mg2+ поступает в виде соли MgАТФ, которая служит субстратом для большинства киназ.
Рис. 163. Путь биосинтеза производных протопорфирина IX. 1 — Mg2+-хелатаза; 2 — S-аденозил-L-метионин: Мg-протопорфирин lX-метилтрансфераза; 3 — циклаза; 4 — 4-винилредуктаза; 5 — оксидоредуктаза; 6 — хлорофилл а-синтаза; 7 — GG-редуктаза.
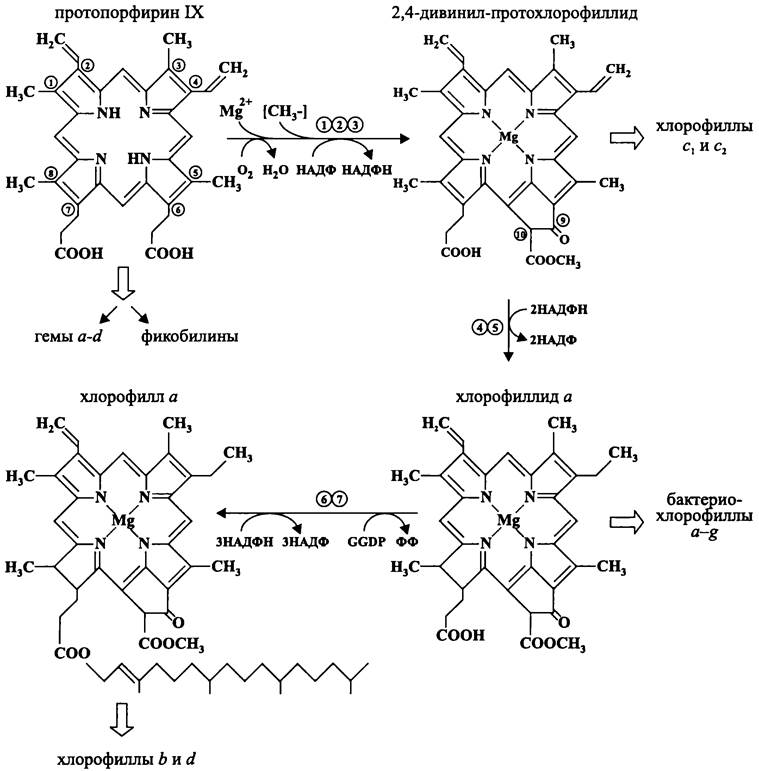
Вторая реакция — это метилирование пропионильной группы в положении 6, что приводит к образованию монометилового эфира Мg-протопорфирина IX. Метильная группа поступает от S-аденозил-L-метионина, который при этом превращается в S- аденозил-гомоцистеин. Катализатором реакции служит S-аденозил-L-метионин:MG- протопорфирин IХ-метилтрансфераза.
Этапы образования циклопентанового кольца V и восстановления винильной группы при II кольце в этильную изучены не так хорошо, как предыдущие.
В частности, предполагается, что замыкание кольца V происходит при участии циклазной системы, по аналогии с механизмом β-окисления жирных кислот. Последовательность событий состоит из дегидрирования, гидратации и окисления гидроксильной группы при β-углеродном атоме остатка пропионовой кислоты, который находится в положении 6 и этерифицирован метанолом. Промежуточными продуктами на пути образования 2,4-дивинил-протохлорофиллида являются 6-метил-β-гидроксипропионил-Мg-протопорфирин IX и 6-метил-β-кетопропионил-Мg- протопорфирин IX. Циклазная система требует присутствия кислорода, НАДФ и катионов Fе2+.
Производными 2,4-дивинил-протохлорофиллида являются хлорофиллы С1 и С2 (см. табл. 13), которыми обладают бурые и другие «хромофитные» водоросли, а также цианобактерии-прохлорофиты Prochloron didemni и Рrochlorococcus marinus.
После восстановления винильной группы в положении 4 из 2,4-дивинил- протохлорофиллида образуется протохлорофиллид. Затем светозависимая НАДФ: протохлорофиллид-оксидоредуктаза (это может быть и «темновой», расходующий АТФ фермент) превращает протохлорофиллид в хлорофиллид а, который имеется у всех оксигенных фототрофных бактерий.
Путем модификации хлорофиллида а образуются бактериохлорофиллы а-g (см. табл. 13, а также раздел 12.7.3). Ими обладают пурпурные бактерии, зеленые бактерии и гелиобактерии.
В 1996 г. группа японских исследователей, возглавляемая Вакао (N. Wakao), обнаружила, что аэробная фототрофная бактерия Acidiphilium rubrum вместо Mg-содержащего бактериохлорофилла а образует Zn-содержащий бактериохлорофилл а. В 2000 г. Вакао и его сотрудники описали вторую бактерию, которая образует Zn-содержащий бактериохлорофилл а — Acidisphaera rubrifaciens. Этот пигмент более стабилен, чем Mg-содержащие (бактерио)хлорофиллы, которые в кислой среде феофитинизируются, т. е. теряют лигандированный катион Mg2+. По-видимому, цинк спонтанно замещает магний на одном из этапов биосинтеза бактериохлорофилла после образования 2,4-дивинил-протохлорофиллида. Однако, как это происходит, еще не ясно.
Для превращения хлорофиллида а в хлорофилл а требуются еще несколько дополнительных реакций. Вначале он этерифицируется полиизопреноидным спиртом геранилгераниолом; при этом роль донора выполняет GGDP. Затем геранилгеранильный остаток последовательно восстанавливается по двойным связям в положениях 6, 10 и 14, в результате чего образуется фитольный хвост. Промежуточными продуктами при восстановлении GG являются 6-дигидро-GG, 6,10-тетрагидро-GG и 6,10,14-гексагидро-GG. Хлорофилл а-синтаза и GG-редуктаза действуют в составе единого комплекса, а восстановителем служит НАДФН.
Производными хлорофилла а являются хлорофиллы b и d. АТФ-зависимое образование хлорофилла b у цианобактерий-прохлорофитов Prochlorococcus mari-
nus, Prochloron didemni и Prochlorothrix sp. осуществляется при помощи хлорофилл а-оксигеназы. Она окисляет метальную группу кольца II в формильную. По- видимому, эта реакция обратима, хотя существование пула взаимно превращающихся молекул хлорофиллов а и b не доказано. Скорее всего, хлорофилл а служит предшественником и для хлорофилла d(см. табл. 13), которым обладает цианобактерия- прохлорофит Acaryochloris marina.