ИММУНОЛОГИЯ - Ройт А. - Мир 2000
Глава 21. Первичная иммунологическая недостаточность
T-КЛЕТОЧНАЯ НЕДОСТАТОЧНОСТЬ
Основные виды Т-клеточной недостаточности приведены на рис. 21.6. Больные с нарушением функции Т-клеток или их отсутствием восприимчивы к оппортунистическим инфекциям. Поскольку функционирование В-лимфоцитов у человека в основном является Т-зависимым, Т-клеточная недостаточность сопровождается также гуморальным иммунодефицитом; иными словами, Т-клеточный дефицит ведет к комбинированной недостаточности как гуморального, так и клеточного иммунитета.

Рис. 21.6. Причины Т-клеточной недостаточности могут быть весьма различными, от полного отсутствия лимфоцитов до недостаточности МНС-антигенов и ферментных дефектов. Все они влияют на функцию Т-клеток, что ведет к комбинированной Т- и В-клеточной недостаточности.
Тяжелый комбинированный иммунодефицит (ТКИД) характеризуется недостаточностью лимфоцитов и недоразвитием тимуса
Наиболее выраженный наследственный дефицит клеточного иммунитета наблюдается у детей с ТКИД, при котором в отличие от Х-АГ уже в раннем возрасте возникают повторные инфекции. У таких детей развивается затяжная диарея, возбудителями которой могут быть ротавирусы или кишечные бактерии, а также пневмония, вызываемая обычно простейшим Pneumocystis caritui. Обычный представитель микрофлоры человека дрожжеподобный гриб Candida albicans дает у них бурный рост в полости рта и на коже (рис. 21.7). Иммунизация таких детей стандартными живыми вакцинами, в частности вакциной против полиомиелита или туберкулеза (БЦЖ), которые, как правило, безвредны для организма, ведет к прогрессирующей инфекции с летальным исходом. Больные ТКИД дети обречены и погибают обычно в первые 2 года жизни, если только им не произвести трансплантацию костного мозга. В этом случае создается лимфоцитарный химеризм, и ребенок может нормально жить и развиваться.
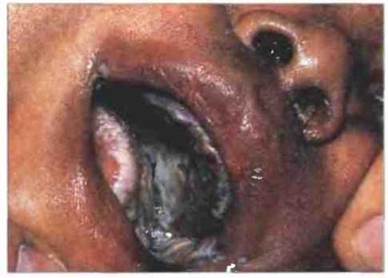
Рис. 21.7. Поражение тканей полости рта Candida albicans при ТКИД. Этот гриб обильно размножается в полости рта и на коже у больных ТКИД.
Кровь больных ТКИД детей содержит очень мало лимфоцитов (<3000/мл). Димфоидная ткань также бедна лимфоцитами или же они в ней вовсе отсутствуют. Тимус у таких детей имеет эмбриональное строение (рис. 21.8); он состоит из энтодермальных клеток стромы, развивающихся в эмбриогенезе из третьего и четвертого глоточных карманов. Стволовые лимфоидные клетки, в норме заселяющие тимус эмбриона к 6 неделе развития (см. гл. 12), в данном случае отсутствуют и тимус не становится лимфоидным органом.
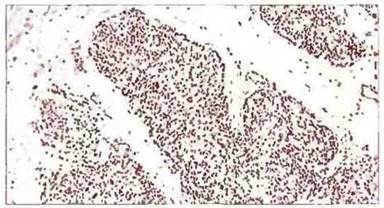
Рис. 21.8. Тимус при ТКИД. Строма не содержит лимфоидных клеток, и тельца Гассаля не обнаруживаются. Орган имеет вид эмбрионального.
ТКИД развивается чаше у мальчиков, чем у девочек (3:1), поскольку более чем в 50 % случаев заболевание обусловлено дефектом гена, расположенного в Х-хромосоме. Этот ген кодирует у-цепь рецептора к ИЛ-2, которая входит также в состав рецепторов к другим интерлейкинам — ИЛ-4, ИЛ-7. ИЛ-II и ИЛ-15. Для созревания Т-клеток особо важное значение имеет взаимодействие ИЛ-7 со своим рецептором. Таким образом, в случае ТКИД, вызванного этим генетическим дефектом, стволовые лимфоидные клетки не могут получать ряд сигналов, необходимых для роста и дифференцировки. В остальных случаях ТКИД связан с дефектами в рецессивных генах других хромосом. Из числа этих больных у половины имеется недостаточность аденозин-дезаминазы (АДА) или пуриннуклеозидфосфорилазы (ПНФ). Дефицит данных ферментов, расщепляющих пурины, ведет к накоплению метаболитов, токсичных для стволовых лимфоидных клеток, дезоксиаденозинтрифосфата (dATP) и дезоксигуанозинтрифосфата (dGТР) (рис. 21.9). Эти метаболиты ингибируют фермент рибонуклеотидредуктазу, необходимую для синтеза ДНК и соответственно для размножения клеток. Поскольку АДА и ПНФ обнаруживаются во всех клетках млекопитающих, возникает вопрос, почему при их недостаточности поражаются только лимфоциты. По-видимому, это связано с относительно низкой активностью 5'-нуклеотидазы в лимфоидных клетках; в других клетках этот фермент компенсирует недостаточность АДА и ПНФ тем, что предотвращает накопление dAMP и dGMP.
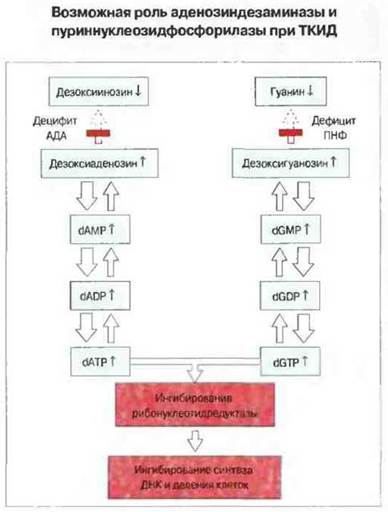
Рис. 21.9. Предполагается, что дефицит аденозиндезаминазы и пуриннуклеозидфосфорилазы ведет к накоплению dATP и dGTP соответственно. Оба метаболита действуют как сильные ингибиторы рибонуклеотидредуктазы - фермента, необходимого для синтеза ДНК.
Оптимальным способом лечения ТКИД служит трансплантация костного мозга, взятого у полностью совместимого по антигенам МНС донора. Обычно таким донором может быть здоровый сибс, однако примерно в 70 % случаев больной ребенок не имеет гистосовместимых братьев и сестер и тогда для пересадки с успехом применяют гаплоидентичный костный мозг одного из родителей. Недавним достижением стало трансфицирование лимфоцитов детей с недостаточностью АДА ретровирусным вектором со вставкой гена АДА первый пример успешного применения «генотерапии».
Недостаточность молекул МНС класса II ведет к дефициту Тх-клеток
Недостаточность экспрессии антигенов МНС класса II антигенпрезентируюшими клетками (макрофагами и В-клетками) наследуется как аутосомно-рецессивный признак, не сцепленный с МНС-локусом в коротком плече хромосомы 6. У больных детей наблюдаются повторяющиеся инфекции с преимущественным поражением желудочно-кишечного тракта. Поскольку развитие Тх-клеток CD4+(Т-хелперов) зависит от положительной селекции с участием молекул МНС класса II в тимусе (см. гл. 12), у детей с недостаточностью этих молекул возникает дефицит Т-лимфоцитов CD4+. Отсутствие Тх-клеток ведет также к недостаточности продукции антител. Дефицит МНС класса II возникает в результате дефектов промоторных белков, которые связываются с 5'-нетранслируемой областью генов класса II.
Синдром Ди Джорджи связан с нарушением развития тимуса в эмбриональном периоде
Эпителий тимуса образуется, как известно, из третьего и четвертого глоточных карманов к 6 неделе эмбрионального развития. Вслед за этим энтодермальную закладку органа заселяют стволовые лимфоидные клетки, дающие начало Т-клеткам. Такое же эмбриональное происхождение имеют паращитовидные железы. Врожденный дефект развития органов, происходящих из третьего и четвертого глоточных карманов, приводит к возникновению синдрома Ди Джорджи. Данный вид недостаточности Т-клеток носит вариабельный характер в зависимости от степени поражения тимуса. Лицо больного ребенка имеет характерные черты (рис. 21.10), широко расставленные глаза (гипертелоризм), низко расположенные уши, укороченный фильтр верхней губы. Описаны также врожденные пороки сердца или дуги аорты, а также гипокальциемические судороги у новорожденных вследствие гипоплазии или аплазии паращитовидных желез.
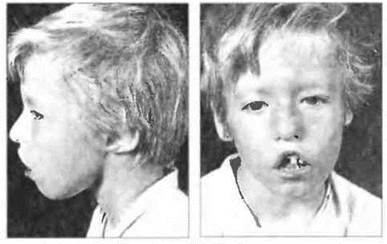
Рис. 21.10. Синдром Ди Джорджи. Характерные признаки - широко расставленные глаза, низко расположенные уши, укороченный фильтр верхней губы. При этом синдроме описаны также врожденные пороки сердца и крупных сосудов.
Наследственная атаксия-телеангиэктазия связана с разрывами хромосом в области генов ТкР и иммуноглобулинов
Атаксия-телеангиэктазия (АТ) наследуется как аутосомно-рецессивный признак. У больных детей в возрасте примерно полутора лет появляются двигательные расстройства, в частности шаткая походка (атаксия). К 6 годам развивается расширение капилляров (телеангиэктазия) конъюнктивы и кожи. Это заболевание сопровождается вариабельной Т-клеточной недостаточностью. Примерно 70 % больных имеет также недостаточность IgA, а у некоторых из них наблюдается еще и дефицит IgG2 и IgG4. Количество и активность циркулирующих Т-клеток резко снижены, в связи с чем наблюдается супрессия Т-клеточного иммунитета. У больных детей развиваются тяжелые инфекции придаточных полостей носа и легких. При цитогенетическом исследовании в клетках обнаруживаются разрывы хромосом, обычно хромосом 7 и 14, в области генов Т-клеточного рецептора (ТкР) и генов, колирующих тяжелые цепи иммуноглобулинов. Клетки больных АТ, как in vivo, так и культивируемые in vitro, высокочувствительны к ионизирующей радиации. Ген, дефектом которого обусловлена АТ, кодирует белок, участвующий в репарации разрывов двух цепочечной ДНК.
Синдром Вискотта-Олдрича обусловлен Т-клеточными дефектами и отклонениями в уровне иммуноглобулинов
Синдром Вискотта-Олдрича (СВО) — это сцепленное с Х-хромосомой иммунодефицитное заболевание мальчиков. Для него характерно уменьшение размеров и резко измененная морфология тромбоцитов наряду с уменьшением их числа (тромбоцитопсния). Заболевание сопровождается экземой, а также гнойными и оппортунистическими инфекциями. В сыворотке крови повышено содержание IgA и IgE, уровень IgG нормальный, a IgM снижен. Функционирование Т-клеток нарушено, и этот дефект клеточного иммунитета прогрессирует. Сканирующая электронная микроскопия позволяет выявить уникальные изменения морфологии Т-клеток больных, отражающие нарушение цитоскелета. Число микроворсинок на поверхности этих клеток снижено по сравнению с нормальными Т-лимфоцитами. Известно, что в ходе кооперации Т- и В-клеток при антителообразовании цитоскелет Т-клетки меняет свою ориентацию или поляризуется в направлении В-клетки. При СВО этого не происходит, и в результате взаимодействие клеток иммунной системы нарушено.