ИММУНОЛОГИЯ - Ройт А. - Мир 2000
Глава 21. Первичная иммунологическая недостаточность
ДЕФЕКТЫ ФАГОЦИТАРНЫХ КЛЕТОК
Фагоцитарные клетки - полиморфноядерные лейкоциты и клетки моноцитарно-макрофагального ряда — выполняют важную роль в защите против гноеродных бактерий и других внутриклеточных микроорганизмов. Выраженная недостаточность полиморфноядерных лейкоцитов (нейтропения) может привести к развитию генерализованной бактериальной инфекции. Особое клиническое значение имеют два генетических дефекта, нарушающих функцию фагоцитов и тем самым вызывающих повышение чувствительности к инфекциям. С этими дефектами связано возникновение тяжелых заболеваний, часто с летальным исходом - хронического гранулематоза и недостаточности адгезии лейкоцитов.
Причина хронического гранулематоза (ХГ) состоит в нарушении механизма восстановления кислорода
У больных ХГ имеется дефект NADP·Н-оксидазы, катализирующей восстановление О2 с образованием ·02-:
![]()
По причине этого дефекта фагоциты больных не способны продуцировать супероксидный кислородный радикал (·02-) и пероксид водорода н поэтому не могут быстро разрушать поглощенные ими клетки бактерий или грибов, в первую очередь каталазообразуюших видов (см. гл. I7). В результате микроорганизмы внутри фагоцитов таких больных остаются жизнеспособными. Персистирующие внутриклеточно микробные антигены вызывают клеточный иммунный ответ и формирование гранулем. У детей, больных ХГ, развиваются пневмония, инфекции лимфоузлов (лимфаденит) и абсцессы в коже, печени и других внутренних органах.
Диагноз ХГ устанавливают по неспособности фагоцитов после стимуляции восстанавливать краситель тетразолиевый нитросиний (ТНС). Поглощаемый фагоцитами, когда они переваривают захваченные частицы, этот краситель, имеющий в окисленной форме бледно-желтую окраску, восстанавливается, присоединяя ион водорода при окислении NADP·Н; при этом в клетках образуются кристаллы ТНС темно-красного цвета. В фагоцитах больных ХГ восстановления ТНС не происходит (рис. 21.14).
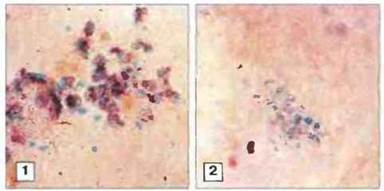
Рис, 21.14. Тест с красителем тетразолиевым нитросиним (ТНС). У здоровых людей в полиморфноядерных лейкоцитах и моноцитах при фагоцитозе образуются высокореакционноспособные метаболиты кислорода (ВМК), при этом желтый ТНС восстанавливается, превращаясь в красно-фиолетовый формазан (1). У больных ХГ образования ВМК не происходит и ТНС сохраняет желтый цвет (2). (Фото любезно предоставлены проф A.R. Hayward.)
Реакция с участием NADP·Н-оксидазы представляет собой сложный процесс, и ферментный комплекс состоит из многих субъединиц. Мембраны покоящихся фагоцитов содержат специфичный для клеток этого типа цитохром b558. Он построен из двух цепей, одна из которых имеет молекулярную массу 91 кДа и кодируется геном в коротком плече Х-хромосомы. Другая цепь, с молекулярной массой 22 кДа, кодируется геном, локализованным в хромосоме 16. При фагоцитозе несколько цитозольных белков фосфорилируются, перемещаются к мембране и связываются с цитохромом b558. Сформированный комплекс действует как фермент NADP·Н-оксидаза, катализируя реакцию окисления NADP·Н, в результате которой образуются высокореакционноспособный кислородный радикал (рис. 21.15). Чаще встречается Х-сцепленная форма ХГ, для которой характерен дефект цепи 91 кДа в составе цитохрома b558. Три других типа ХГ являются аутосомно-рецессивными расстройствами и обусловлены дефектами другой цепи (22 кДа) цитохрома b558 или одного из двух белков, р47phox или р67phox (phox — сокращение от англ. phagocytic oxidase).
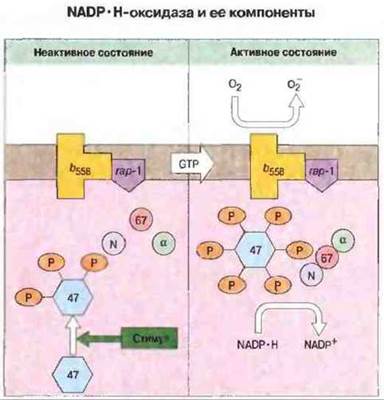
Рис. 21.15. Согласно существующим представлениям о природе данной NADP·H-оксидазы, в отсутствие активирующего стимула некоторые из компонентов этого фермента локализованы в мембране (цитохром b558, и, возможно, rар-1), тогда как другие находятся в цитозоле (p47phox, р67рhox, NADP·Н-связывающий компонент N и предполагаемый четвертый компонент α). В ответ на стимул, индуцированный фагоцитозом, происходит сборка комплекса из цитозольных элементов, который перемещается затем к мембране. Возможно, этот процесс опосредован фосфорилированием (Р) р47phox. Как только цитозольные компоненты связываются с мембранными, оксидаза становится каталитически активной и р47phox фосфорилируется далее. При различных формах ХГ обнаружены дефекты генов, кодирующих различные компоненты этой оксидазы. (По Smith R.M., Curnutte J Т., 1991 Molecular basis of chronic granulomatous disease. Stood 77 (4): 673-86, с разрешения).
Недостаточность адгезии лейкоцитов (НАЛ) обусловлена дефектами в генах интегринов
Для поглощения микроорганизмов фагоцитарными клетками решающее значение имеет мембранный рецептор фагоцитов, который связывается с С3bi на поверхности опсонизированных микробных клеток. У больных, страдающих НАД, этот рецептор интегрин, названный рецептором 3 комплемента (CR3), — отсутствует, вследствие чего они подвержены тяжелым бактериальным инфекциям, особенно часто поражающим полость рта и желудочно-кишечный тракт.
Молекула CR3 построена из двух полипептидных цепей: α-цепи с молекулярной массой 165 кДа (CD11b) и β-цепи с молекулярной массой 95 кДа (CD18). При НАД имеется генетический дефект β-цепи, которую колирует ген в хромосоме 21. Ту же самую β-цепь содержат два других интегриновых белка - лейкоцитарный функциональный антиген (LFA-1) и р150,95 (см. гл. 5). Хотя каждый из этих белков имеет свою a-цепь (CD11a и CD11c соответственно), оба она при НАД дефектны. Молекула LFA-1 играет важную роль в клеточной адгезии; она взаимодействует с молекулой 1 межклеточной адгезии (ICAM-1) на поверхности эндотелиальных клеток и других клеточных мембранах. Вследствие дефекта LFA-1 фагоциты больных НАД не способны прикрепляться к сосудистому эндотелию и поэтому не могут мигрировать из кровеносных сосудов в область внедрения инфекционного агента. У больных не происходит образования локальных гнойных очагов, в связи с чем бактерии имеют возможность быстро распространяться по организму.
Вопросы для размышления
■ Клиническая картина при Х-сцепленной агаммаглобулинемии неотличима от проявлений наследственной недостаточности третьего компонента комплемента (С3): и те и другие больные имеют повышенную чувствительность к гнойным инфекциям. Как это объяснить?
■ У лиц с недостаточностью антигенов МНС класса II отсутствуют Т-клетки CD4+. В редких случаях наблюдается недостаточность МНС-антигенов класса I. Недостаточность какой субпопуляции Т-клеток должна быть у этих больных?
■ Лица с недостаточностью компонентов комплемента С1, С2, С4 и ингибитора С1 могут быть не столь восприимчивы к гнойным инфекциям, как больные с дефицитом С3. В чем причина этого различия?
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Conley М.Е. 1991. Molecular approaches to analysis of X-linked immunodeficiencies. Ann. Rev. Immunol. 10: 215.
Cumutte J.T., Orkin S.H.. Dinauer M.C. Genetic disorders of phagocyte function. In: Stramatoyannopoulos G., Nienhuis A.W., Majerus P.W., Varmus H. (eds.). The Molecular Basis of Blood Diseases. Philadelphia: PA Saunders; 1994: 443.
Rosen F.S., Cooper M.D., Wedgwood R.J.P. 1995. The primary immunodeficiencies. N. Engl. J. Med. 333: 43.
Rosen F.S., Seligman M. (eds.). Immunodeficiencies. Switzerland: Harwood Academic Publishers GmbH. 1993.
Von Andrian U.H., Berger E.M., Chambers, J.D. et al. 1993. In vivo behaviour of neutrophils from two patients with distinct inherited leukocyte adhesion deficiency syndromes. J. Clin. Invest. 91: 2893.