Аминокислоты, пептиды и белки - Дэвени Т., Гергей Я. 1976
Некоторые методологические вопросы аналитического исследования белков
Методологические вопросы анализа аминокислотной последовательности белков
Для расшифровки аминокислотной последовательности белков и полипептидов, как и для любого другого структурного анализа, к объектам исследования предъявляется ряд требований. Подлежащий анализу материал должен быть однородным, а его основные физические параметры и прежде всего молекулярный вес и точный аминокислотный состав должны быть известны. Выполнение этих условий, однако, зачастую ставит перед исследователями довольно трудные задачи.
Проблема гомогенности белков в свое время решалась относительно просто с помощью традиционных методов анализа. Препарат считали гомогенным, если он не делился на фракции при электрофорезе или при ультрацентрифугировании. В настоящее время эти критерии гомогенности утратили свое значение. В нашем распоряжении появились более чувствительные методы исследования и стали обязательными более строгие показатели гомогенности. Ионообменная хроматография и электрофорез в геле являются сейчас наиболее чувствительными методами выявления так называемой микрогетерогенности белковых препаратов, какова бы ни была ее природа. Термин “микрогетерогенность” предложили Синг [82] в 1943 г. и Колвин с сотр. [6] в 1954 г. Из методов электрофореза в геле наиболее чувствительным для анализа микрогетерогенности белков оказался вертикальный диск-электрофорез. При использовании этого метода требуются весьма малые количества необходимого для исследования материала, с его помощью можно одновременно анализировать большое число образцов и к тому же он отличается высокой разрешающей способностью. Диск-электрофорезом можно обнаружить микрогетерогенность препарата даже в том случае, если компоненты имеют идентичную аминокислотную последовательность, но различаются по конформации молекул. Этот метод позволяет выявлять также самые незначительные химические различия.
Высокая разрешающая способность хроматографии с использованием ионообменных целлюлоз, декстрана и других материалов делает этот метод наиболее подходящим для выделения гомогенных белковых препаратов.
В последнее время появилась возможность определять аминокислотный состав белков с помощью автоматических аминокислотных анализаторов. Когда в 1948 г. Мур и Стейн [55] в дополнение к классическим методам органической химии, а также манометрическому и бактериологическому анализу ввели ионообменную хроматографию, наступил поворотный момент в развитии химии аминокислот. В основу работы созданных сотрудниками Рокфеллеровского института современных автоматических аминокислотных анализаторов была положена ионообменная хроматография. Принцип работы этих приборов заключается в следующем. Исследуемый белок гидролизуют, затем гидролизат подвергают хроматографии на смоле типа дауэкс 50 х8 в Na-форме. Элюирование производят с помощью непрерывной подачи буферного раствора. Выходящий из колонки элюат попадает в пластмассовую ячейку особой формы, где он смешивается с раствором нингидрина. Подачу нингидрина осуществляет специальный насос, работающий синхронно с насосом, подающим буферный раствор на колонку. Затем смесь элюата с нингидрином проходит через тефлоновый капилляр, который погружен в кипящую баню. В этих условиях в растворах происходит нингидриновое окрашивание, интенсивность которого измеряется в проточной кювете спектрофотометрически. Поглощение света регистрируется самописцем. Применение сферических смол [80] позволило сократить время исследования одного образца примерно в четыре раза, а использование особых ячеек сделало вполне допустимыми для анализа очень малые количества исследуемого вещества — порядка 0,01—0,05 мкмоля [38]. Введение одноколоночной процедуры значительно упрощает метод [9, 29, 43, 60]. С помощью этой методики в одной и той же пробе можно определить кислые, нейтральные и основные аминокислоты, что не только экономит исследуемый материал, но и повышает точность и сокращает время исследования. Работая на стандартном аминокислотном анализаторе и пользуясь некоторыми модификациями известных методов, можно полностью закончить анализ одного вещества в течение 3 ч [9].
Определение аминокислотной последовательности осуществляется по принципу “собирания мозаики”. Предположим, что мы имеем вещество с последовательностью А. В.С. D.E. F. G. Н. I. J. К. L. М. N и можем воспользоваться двумя методами специфического расщепления пептидных связей. Один из методов разрывает связь
... F. G ..., a другой — связь ... H. I... Применим оба метода расщепления к нашей модели. Первый метод даст два пептида: А. В. С. D. Е. F. и G. Н. I. J. К. L. М. N. Второй метод даст также два пептида: А. В. С. D. Е. F. G. Н. и I. J. К- L. М. N. Последовательность исходного пептида определяют на основании анализа четырех полученных фрагментов. Для этого нужно определить их аминокислотный состав, а также N- и С-концевые группы. Аминокислотную последовательность этих малых пептидов удается определить путем поэтапного расщепления, дальнейшего частичного гидролиза и определения концевых групп получившихся более мелких фрагментов. При этом возникает возможность выявить “перекрывающиеся” последовательности пептидных фрагментов. Например, часть G… Н... пептида, полученного первым методом расщепления, имеет такую же последовательность, как и С-концевая часть фрагмента ... F. G. Н.— одного из пептидов, полученных вторым методом. По принципу “собирания мозаики” мы может восстановить структуру исходного пептида на данном участке.
Очевидно, чем крупнее молекула анализируемого белка, тем сложнее проблема отыскания перекрывающихся последовательностей, причем одним из наиболее важных моментов в определении последовательности является специфический гидролиз.
Значительное затруднение при расшифровке последовательности аминокислот может возникнуть, если в молекуле анализируемого белка присутствуют остатки цистеина или цистина. При окислении цистеина образуются S—S-мостики, которые не только являются причиной ошибочных выводов, но и препятствуют дальнейшему анализу, так как содержащие их белки и полипептиды весьма устойчивы к ферментативному расщеплению. Поэтому до проведения анализа рекомендуется избавляться от S—S-мостиков и предотвращать спонтанное окисление свободных SH-групп. Кроме того, следует иметь в виду возможность SH/S—S-обмена. Если в реакционной смеси одновременно присутствуют свободные SH-группы и S—S-мостики, в ней могут происходить перестройки, при которых связанные S—S- мостиком пары пептидов обмениваются своими партнерами:
R1—S—S—R2 + R3—S—S—R4⇄ R1-S—S—R3 + R2—S-S-R4.
SH- и S — S-группы элиминируют с помощью количественной и необратимой реакции окисления белков надмуравьиной кислотой. В результате этой реакции цистеин или цистин превращается в цистеиновую кислоту [86].
![]()
При окислении надмуравьиной кислотой триптофан разрушается, а значительная часть тирозина при последующем гидролизе НСl превращается в хлорированный тирозин [85]. При низкой температуре окисление проходит более мягко, причем в этих условиях количественный характер образования цистеиновой кислоты сохраняется [35].
Разрыв S — S-связей можно вызвать также с помощью реакции восстановления. Если в качестве восстановителей используются тиосоединения, то в ходе реакции возникают свободные реакционноспособные SH-группы, которые могут стать помехой дальнейшему анализу. Поэтому после реакции восстановления их необходимо блокировать. Лучшим способом блокирования является карбоксиметилирование. Схема реакции такова:
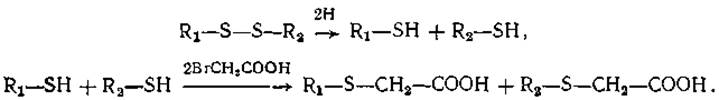
В результате образуется производное карбоксиметилцистеина. Если в этой реакции использовать 14С- Вr- или 14С-I- ацетат, то, помимо разрыва S—S-связей и блокирования возникающих SH-гpyпп, можно радиоактивно пометить пептиды, содержащие цистеиновые остатки.
Одним из вариантов восстановления является реакция сульфитолиза. В присутствии Сu2+ при щелочном pH соединения, имеющие S—S-связи (или свободные SH-группы), под влиянием солей сернистой кислоты превращаются в S-сульфонаты. Реакция происходит следующим образом:
![]()
В структурном анализе белков реакция сульфитолиза никогда широко не использовалась, и в настоящее время ее почти не применяют [5, 11, 14, 33, 59].
Одна из основных стадий расшифровки аминокислотной последовательности состоит в частичном расщеплении молекул исследуемого белка. Используемые для этой цели методы частичного гидролиза или специфического химического расщепления, кроме максимально возможной специфичности, должны отвечать следующим требованиям: 1) в ходе реакции аминокислоты не должны разрушаться; 2) специфичность реакции, т. е. расщепление в строго определенном месте, должна быть заранее известна; 3) в ходе расщепления не должно возникать побочных реакций, таких, как перенос ацильной группы, транспептидирование или образование случайных пептидов.
Методы частичного гидролиза используются широко, но почти все они лишены специфичности и поэтому пригодны только для получения мелких фрагментов, например на конечных этапах расщепления больших пептидов. Самым старым из методов неспецифического, частичного гидролиза является частичный кислотный гидролиз. Сэнгер и Туппи [72] применили этот метод при расшифровке структуры В-цепи инсулина. Выделив и проанализировав не менее 60 пептидов, им удалось расшифровать только четыре участка цепи, включающих всего 19 остатков аминокислот. В частичных кислотных гидролизатах, помимо пептидов, встречается до 25% свободных аминокислот [54]. В ходе кислотного гидролиза полностью разрушается триптофан [53] и в значительной степени повреждаются оксиаминокислоты [65].
Из методов неспецифического ферментативного расщепления чаще всего применяется гидролиз пепсином, папаином, бактериальными или грибковыми протеазами. Все три типа ферментов дают гидролизаты, представляющие собой сложную смесь мелких пептидов. В связи с этим их лучше использовать на конечных этапах расщепления крупных пептидов, полученных методами специфического гидролиза. Общий обзор методов ферментативного гидролиза сделал Хилл [34]; подробные сводки о действии пепсина, папаина и бактериальных протеаз опубликовали Бовей и Янари [2], Смит и Кщммель [78] и Хагихара [28] соответственно.
Специфический ферментативный гидролиз можно провести в присутствии трипсина и химотрипсина. Эти ферменты по современным представлениям обладают наибольшей специфичностью расщепления, а их препараты, имеющиеся в продаже, отличаются высокой чистотой.
Трипсин гидролизует пептидные связи, образуемые основными аминокислотами, т. е. связи, в которых участвуют остатки лизина и аргинина. Пептидные связи Лиз-Про и Арг-Про устойчивы к гидролизу. Частичной устойчивостью к трипсиновому гидролизу обладают также некоторые другие пептидные связи, например в структуре ... Лиз-Лиз-Х... связь Лиз-Лиз или связи в пептидах Арг-Арг, Арг-Лиз и Лиз-Арг. “Скопление” основных аминокислот в определенных участках пептида обусловливает частичную устойчивость его к гидролизу. То же самое справедливо и для пептидных связей Лиз-Глу и Арг-Глу.
Примесь химотрипсина в препарате трипсина может в значительной степени исказить специфичность трипсинового гидролиза, поэтому крайне важно избегать загрязнения трипсина химотрипсином. Загрязненный препарат трипсина можно очистить хроматографией [84]; химотрипсин, содержащийся в препарате, можно инактивировать кислотой или недавно полученными синтетическими ингибиторами.
Благодаря специфичности триптического гидролиза им можно “управлять”, внося те или иные изменения в структру гидролизуемых соединений. Например, по мере аминоэтилирования свободных SH-групп увеличивается число NH2-гpyпп и, следовательно, увеличивается количество пептидных связей, чувствительных к трипсину, т. е. в гидролизате оказывается больше триптических пептидов [48, 63]. Если же, наоборот, обратимо или необратимо заблокировать NH2-гpyппы лизина, то трипсин сможет гидролизовать только те пептидные связи, в которых участвует аргинин. В этом случае в гидролизате обнаруживаются крупные аргинило- вые пептиды. При обратимом блокировании остатков лизина, например путем трифторацетилирования, после выделения аргиниловых пептидов их можно деблокировать, а затем подвергнуть триптическому гидролизу; при этом будут расщепляться только пептидные связи лизина.
Новейшим методом обратимого блокирования ε-NH2-гpyпп можно считать реакции малеинирования [47] ицитраконилирования [13]. Оба реагента, участвующие в этих реакциях, не только блокируют аминогруппы, но и увеличивают растворимость крупных фрагментов, полученных при триптическом гидролизе. Благодаря обратимости обеих реакций ацилирующие реагенты подобного типа могут с успехом применяться для расшифровки первичной последовательности.
Изложенный выше метод “управляемого протеолиза” играет существенную роль в анализе аминокислотной последовательности высокомолекулярных белков.
Химотрипсин расщепляет больше пептидных связей, чем трипсин. При кратковременном гидролизе в течение 2—3 ч фермент расщепляет пептидные связи, в которых участвуют остатки тирозина, фенилаланина и лейцина. И в этом случае полную устойчивость к гидролизу сохраняют пептиды пролина. “Скопления” ароматических аминокислот, например структуры, подобные ... Фен-Фен ... или... ... Тир-Фен ...и т. д., обладают частичной устойчивостью к гидролизу. Однако с увеличением продолжительности гидролиза происходит разрушение пептидных связей многих типов. Подробный обзор по химотрипсину был сделан Деснуэллем [7]; сведения о специфичности химотриптического гидролиза можно найти в обзоре Хилла [34].
Специфическое химическое расщепление пептидных связей можно осуществить с помощью двух окисляющих агентов. Один из них, N-бромсукцинимид, вызывает расщепление пептидной связи, в которой участвует остаток триптофана, с одновременным разрушением этого остатка и его окислением [64]. Успешное использование этого метода удалось лишь в нескольких случаях. Помимо изучения структуры полипептида, выделенного из вируса табачной мозаики, N-бромсукцинимид был использован при анализе последовательности низкомолекулярного глюкагона [58] и N-kohцевой последовательности гемоглобина [79]. Существенный недостаток этого метода заключается в том, что N-бромсукцинимид расщепляет не все пептидные связи, образуемые остатками триптофана. В реакциях с одним и тем же препаратом белка могут быть окислены от 2 до 80% этих связей. Более того, некоторые белки, например цитохром с, оказываются полностью устойчивыми к действию N- бромсукцинимида.
Другой окислитель, бромциан, — более надежный реагент [26]. Он разрушает пептидные связи, в которых участвует остаток метионина. Механизм этой реакции подобен расщеплению иодацетамидом [45]. Окисление бромцианом широко применяют как специфичный метод для получения крупных пептидных фрагментов. Весьма успешно он был использован наряду с другими методами при расшифровке аминокислотной последовательности рибонуклеазы [27], миоглобина [17], трипсиногена [36] и альдолазы [42, 68,69].
Даже при специфическом ферментативном или химическом расщеплении белка или полипептида среднего молекулярного веса получается довольно сложная смесь пептидов. Существует набор разнообразных методических приемов фракционирования этой смеси и очистки отдельных компонентов. Вряд ли можно предложить какую-то одну схему фракционирования, которая была бы равным образом применима ко всем белковым гидролизатам. Первым этапом обычно является предварительное фракционирование в соответствии с зарядом или размером пептидов. За ним следует уже окончательная очистка пептидов с помощью электрофореза, ионообменной или иной хроматографической процедуры.
Предварительное разделение смеси пептидов в соответствии с их электростатическим зарядом можно осуществить с помощью свободного ионофореза (или многокамерного электрофореза), который успешно применяли Сэнгер и Туппи [72]. Однако этот метод в настоящее время полностью вытеснен гель-фильтрацией, т. е. разделением в соответствии с размерами молекул. После появления первых работ в этом направлении [44, 83] чрезвычайно полезный метод гель-фильтрации был разработан Поратом и Флодиным [61]; позднее Флодин теоретически обосновал его [20].
При гель-фильтрации наиболее употребительны два типа гелей: гели, полученные путем ферментирования декстрана (сефадексы), и синтетические гели акриламида (биогели). Акриламидные гели обладают низкой адсорбционной способностью, поэтому при равной разрешающей способности для них характерен больший выход фракционируемого вещества, чем для гелей декстрана.
После предварительного фракционирования дальнейшую очистку смеси пептидов удобно производить с помощью электрофореза на бумаге. Существует четыре разновидности этого метода: 1) вертикальный электрофорез на бумаге без охлаждения [51]: 2) вертикальный электрофорез на бумаге с охлаждением толуолом [50]; 3) горизонтальный электрофорез на бумаге с охлаждением типа “сэндвич” [25]; 4) горизонтальный электрофорез на бумаге с охлаждением подложки [10].
С помощью горизонтального электрофореза на бумаге можно проводить анализ смеси пептидов методом “отпечатков пальцев” и выделять микроколичества различных фракций. Для решения этих задач удобен электрофорез на бумаге в двух буферных растворах, который позволяет в одноэтапном эксперименте четко разделить кислые и основные компоненты смеси [8]. После гидролиза средней продолжительности в реакционной смеси обычно присутствует смесь кислых и основных пептидов. Одновременная подача двух разных буферных растворов способствует более полному разделению таких пептидов.
Применение при электрофорезе летучих буферов позволяет сразу же после высушивания бумаги элюировать разделенные пептиды и проводить их дальнейшее фракционирование.
Из летучих буферов чаще всего используют системы пиридин — уксусная кислота и уксусная кислота — муравьиная кислота с величинами pH 6,5; 3,5 и 1,9.
В ходе очистки вслед за электрофорезом при разных значениях pH для разделения пептидов применяется хроматография на бумаге.
Общепринято определять пептиды с помощью нингидриновой реакции. Добавление Cd2+ к раствору нингидрина не только увеличивает ее чувствительность, но и повышает стабильность окрашивания. N-ацильные пептиды можно выявлять в реакции с хлором [67]. Некоторые аминокислоты выявляются реакциями специфического окрашивания, например гистидин — реакцией Паули, аргинин — реакцией Сакагуши. С помощью этих же реакций можно обнаруживать и пептиды, содержащие гистидин и аргинин.
Если электрофорез и хроматографию на бумаге можно отнести к микропрепаративным методам, то метод разделения смеси пептидов с помощью ионообменной хроматографии следует, пожалуй, считать макропрепаративным. Его главное преимущество заключается в том, что он позволяет обрабатывать большие количества материала и получать несомненно большие выходы фракций. Применение при ионообменной хроматографии летучих буферов дает возможность избежать трудоемкой процедуры обессоливания, которая осложняла ранее предложенные методики [49, 92]. Большим достижением в области ионообменной хроматографии является введение сферических смол. Их применение способствует увеличению скорости потока фракционируемых веществ через колонку и значительно сокращает продолжительность препаративного разделения. Сферические смолы в автоматических аминокислотных анализаторах обеспечивают воспроизводимую сравнительную хроматографию пептидов с высокой разрешающей способностью, т. е. позволяют автоматически проводить анализ методом “отпечатков пальцев”.
Ключевая роль в расшифровке последовательности принадлежит определению концевых аминокислот, имеющих либо свободную NH 2-группу на N-конце, либо свободную СООН-группу на С-конце пептида. Именно поэтому классический метод Сэнгера явился значительным достижением в белковой химии; его принцип заключается в следующем. Анализируемый белок или пептид в очень мягких условиях обрабатывают 2,4-динитрофторбензолом (ДНФБ). В результате происходит динитрофенилирование N-концевых аминогрупп, а также некоторых реакционноспособных боковых цепей, имеющих следующие группы: ε-NH2, фенольные — ОН, имидазольные —NH и — SH. После этого динитрофенильные (ДНФ) производные белков или пептидов подвергают кислотному гидролизу и экстрагируют эфиром а-ДНФ-аминокислоты, занимавшие в белке или пептиде N-концевое положение. После экстракции в гидролизате остаются только водорастворимые ДНФ-аргинин и ДНФ-лизин. Затем хроматографическим анализом эфирного экстракта точно устанавливают концевую аминокислоту, используя колоночную [1,62], бумажную [46, 66], а также тонкослойную хроматографию [3].
Дансильный метод, который разработали Грей и Хартли в 1963 г. [24], во многих отношениях сходен с динитрофторбензольным методом. Основной реагент в дансильном методе — диметиламинонафтилсульфохлорид. Дансильная группировка присоединяется к тем же группам пептида, что и ДНФБ, но благодаря интенсивной флуоресценции дансильные производные могут быть обнаружены в очень малых количествах (до 10-4 мкмолей вещества).
Другим методом определения N-концевой аминокислоты служит деградация фенилизотиоцианатом по Эдмону [15, 16]. Фенилизотиоцианат превращает N-концевые аминокислоты в фенилтиокарбамильные производные, отщепляющиеся в виде производных фенилтиогидантоина в условиях, при которых не повреждаются другие пептидные связи. Этот метод позволяет осуществить постепенную деградацию полипептидов и пептидов. Отщепившийся аминокислотный остаток можно идентифицировать хроматографией фенилтиогидантоиновых производных [76] или сопоставлением аминокислотного состава укороченных пептидов и исходного материала. В последнее время реакцию Эдмана обычно комбинируют с дансильным методом, так как в таком варианте для определения аминокислоты, занявшей концевое положение, требуется очень малое количество деградируемого пептида.
N-концевую последовательность можно также определить с помощью фермента аминополипептидазы, который специфически гидролизует N-концевые пептидные связи (т. е. связи, образуемые аминокислотой со свободной a-NH2-группой) [77, 81]. Освободившиеся после гидролиза аминокислоты идентифицируют хроматографически.
Подобным образом с помощью карбоксипептидаз А и В можно определить С-концевые аминокислоты и С-концевую последовательность. Если в С-концевом положении находится основная аминокислота, то ее можно отщепить лишь карбоксипептидазой В [21].
Вслед за первыми работами Сэнгера в последние годы удалось расшифровать первичную структуру многих полипептидов и белков. После определения аминокислотной последовательности инсулина были расшифрованы последовательности рибонуклеазы, полипептида из вируса табачной мозаики, нескольких препаратов цитохрома с, различных цепей гемоглобина, ингибитора трипсина, химотрипсиногена и химотрипсина, трипсина, глицеральдегидфосфатдегидрогеназы и т. д. Вместе с последовательностями некоторых пептидных гормонов эти данные вошли в “Атлас структуры и аминокислотной последовательности белков”, который впервые был опубликован в 1966 г. и затем неоднократно переиздавался1.
Второе издание этого атласа, опубликованное в 1967—1968 гг., вдвое больше первого и представляет собой исключительно ценную сводку результатов работ по расшифровке последовательности. Там приведены последовательности цитрохрома с, ферредоксина, цепей гемоглобина, миоглобина, различных иммуноглобулинов и их L-цепей, трипсиногена, трипсина, химотрипсиногена, химотрипсина, субтилизина, а-лактальбумина, триптофан-синтазы, лизоцимов, рибонуклеазы, ингибитора трипсина, пептидных гормонов, ядов, токсинов, вирусных белков и т. д. Включая видовые вариации, число белков с расшифрованной структурой достигает почти двухсот.
Для решения определенных биологических проблем вовсе не обязательна расшифровка всей последовательности изучаемого белка. Например, при выяснении связи между структурой и функцией или при анализе видовой специфичности, а также при решении ряда других проблем требуется выяснить последовательность определенной части структуры того “ответственного” (marked) пептида, который непосредственно участвует в рассматриваемом процессе. В зависимости от изучаемой функции понятие “ответственный” пептид может быть достаточно широким. Это либо активный центр, либо участок связывания, либо, наконец, локус специфичности.
Выяснить локализацию “ответственного” пептида далеко не просто. Природа редко наделяет интересную с точки зрения экспериментатора структуру какой-либо меткой. Однако такая метка имеется, например, у гем-связывающего пептида цитохрома с. Часть пептида, непосредственно участвующая в осуществлении биологической функции, ковалентно соединена тиоэфирной связью с простетической группой цитохрома. Эта стабильная связь облегчает выделение и анализ этого пептида. Туппи и сотр. [87—91] удалось сравнить последовательности гем-связывающих пептидов, выделенных из цитохромов с разного видового происхождения. Некоторые из этих данных приведены в табл. 6
1 Одно из последних изданий опубликовано в 1972 г. (том 5) под редакцией Дейхоф; приложение к нему вышло в свет в 1973 г. (см. список литературы). — Прим. ред.
Таблица 6 Гем-связывающие пептиды из цитохромов с разного видового происхождения

Как видно из данных табл. 6, цитохромы даже очень отдаленных видов имеют значительное сходство гем-связывающих пептидов, особенно в той части, которая включает последовательность ... Цис-Гис-Тре ... . Вместе с результатами более поздних исследований эти данные подтверждают справедливость существующего в химии белков принципа “сходная структура—сходная функция”.
Другой весьма примечательный пример изучения “ответственных” пептидов можно найти в истории исследования эстераз. Янсен и др. [39] обнаружили, что в эквимолярных концентрациях диизопропилфторфосфат (ДИПФФ) способен необратимо подавлять активность некоторых протеаз и эстераз. Из гидролизатов этих ферментов удалось выделить и расшифровать структуру тех пептидов, с которыми связывается радиоактивный ингибитор. Результаты этой расшифровки представлены в табл. 7. У всех сравниваемых ферментов аминокислотные остатки, расположенные вблизи заблокированной ДИПФФ боковой цепи, содержащей серин, оказались идентичными или сходными. Это сопоставление также подтвердило справедливость принципа “сходная структура — сходная функция”. В химии белков этот принцип следует принимать с известной оговоркой: аналогичной структуре не всегда соответствует тождественная функция, а чаще всего похожая или генетически близкая. Сходные структуры могут быть генетически близкими и возникать, например, от общего предшественника путем замены аминокислот в результате изменения одного основания в их кодоне.
Обработка дегидрогеназ радиоактивным иод ацетатом и последующее фракционирование гидролизата позволяют определить последовательность аминокислот, расположенных вблизи реакционноспособных SH-групп в молекуле этих ферментов. Этим способом была расшифрована последовательность вблизи активного центра глицеральдегидфосфатдегидрогеназы и алкогольдегидрогеназ [30, 31,47]. Аналогичным образом была определена последовательность того же участка лактатдегидрогеназы [23, 37]. Результаты этих исследований представлены в табл. 8.
Таблица 7 Пептиды эстераз, содержащие серия
Фермент |
Последовательность |
Источник данных |
Химотрипсин |
Гли-Асп-Сер-Гли-Гли |
[32, 57, 73] |
Трипсин |
Гли-Асп-Сер-Гли-Про |
[12] |
Тромбин |
Асп-Сер-Гли |
[22] |
Эластаза |
Асп-Сер-Гли |
[56] |
Бутирилхолинэстераза |
Гли-Глу-Сер-Ала-Гли |
[40] |
Ацетилхолинэстераза |
Гли-Сер-Ала |
[70, 74] |
Алиэстераза из печени лошади |
Гли-Глу-Сер-Ала-Гли |
[41] |
Субтилизин |
Гли-Тре-Сер-Мет-Ала |
[71 |
Протеаза из Aspergillus orizae |
Тре-Сер-Мет-Ала |
[70] |
Фосфоглюкомутаза |
Тре-Ала-Сер-Гис-Асп |
[52] |
Фосфорилаза |
Глн-Иле-Сер-Вал-Арг |
[19] |
Щелочная фосфатаза |
Тре-Асп-Сер-Ала-Ала |
[18, 75] |
Структурные аналогии, указанные в табл. 8, дают возможность разделить дегидрогеназы на две группы. В первую группу входят лактат- и алкогольдегидрогеназы. Вторую группу образуют оба вида триозодегидрогеназ. Наши сегодняшние знания о функции этих ферментов, несмотря на противоречивость, в известной мере подтверждают такую классификацию. Например, довольно определенно показано, что реакционноспособные SH-группы включенных в таблицу двух триозодегидрогеназ сходным образом принимают участие в образовании промежуточного комплекса — ацилированного фермента. Обе триозодегидрогеназы в соответствующих участках структуры аналогичны одна другой, но не имеют сходства с ферментами первой группы. Таким образом, структурной аналогии действительно сопутствует функциональное сходство.
Таблица 8 “Активные” пептиды, содержащие цистеин, в различных дегидрогеназах
Фермент |
Последовательность |
Лактатдегидрогеназа сердечной ткани цыпленка |
Сер-Гли-Тля-Цис-Асн-Лей-Асп |
Алкогольдегидрогеназа дрожжей |
Тре-Гли-Вал-Цис-Гис-Тре-Асп |
Алкогольдегидрогеназа печени |
Сер-Гли-Иле-Цис-Арг-Сер-Асп |
Глииеральдегидфосфатдегидрогеназа |
Асн-Ала-Сер-Цис-Тре-Тре-Асн |
Глицерофосфатдегидрогеназа |
Вал-Асп-Тре-Цис-Сер-Гли |
Цитированная литература
1. Bailey К., Biochem., J., 49, 23 (1951).
2. Bovey F. A., YanariS. S., in: Boyer P., Lardy H., Myrbäck K., The Enzymes, Academic Press, New York (1960).
3. Brenner AL, Niederwieser A., Pataky G., Experientia, 17, 145 (1961).
4. Butler P. J. E., Harris J. I., Hartley B. S., Leberman R., Biochem. J., 112, 679 (1961).
5. Cecil R., Loening U. E., Biochem. J., 76, 146 (1960).
6. Colvin J., Smith D. В., Cook W. AL, Chem. Rev., 54, 687 (1954).
7. Desnuelle P., in: Boyer P., Lardy H., Myrbäck K., The Enzymes, Academic Press, New York (1966).
8. Devenyl T., Magyar K.em, Folyóirat, 69, 538 (1963).
9. Devenyl T., Acta Biochim. Boiphys. Acad. Sci. Hung., 3, 429; 4, 297 (1968, 1969).
10. Devenyi T., Sajgó AL, Horvath E., Szozenyi B., Magyar Kem, Folyóirat, 70, 123 (1964).
11. Dinh van Hoang, Rovery AL, Deśnuelle P., Biochim. Biophys. Acta, 69, 188 (1963).
12. DixonG. H., KauffmannD. L., Neurath Al., J. Biol., Chem., 233, 1373(1958).
13. Dixon H. B. F., Perham R. N., Biochem. J., 109, 312 (1968).
14. Dlouha V., Keil В., Sorm F., Coll. Czechoslov. Chem. Commun, 28, 2969 (1963).
15. Edman P., Acta Chem. Scand., 4, 283 (1950).
16. Edman P., Acta Chem. Scand., 7, 700 (1953).
17. Edmundson A. B., Nature, 198, 354 (1963).
18. Engström L., Biochim, Boiphys. Acta, 56, 606 (1962).
19. Fisher E. H., Graves D. J., Crittenden E. R. S., Krebs E. G., J. Biol. Chem. 234, 1698 (1959).
20. Flodin P., J. Chromatogr., 5, 103 (1961).
21. Gladner J. A., Folk J. E., J. Biol. Chem., 231, 393 (1957).
22. Glander J. A., Laki K., J. Am., Chem., Soc., 80, 1263 (1958).
23. Gold A. H., Segal H., Biochemistry, 4, 1506 (1965).
24. Gray W. R., Hartley R. S., Biochem., J, 89, 379 (1963).
25. Gross D., Nature, 176, 72 (1955).
26. Gross E., Witkop B., J. Am. Chem. Soc., 83, 1510 (1961).
27. Gross E., Witkop B., J. Biol. Chem., 237, 1856 (1962).
28. Hagihara B., in: Boyer P., Lardy H., Myrbäck K., The Enzymes, Academic Press, New York, 4, 193 (1960).
29. Hamilton P. B., Anal. Chem., 35, 2055 (1963).
30. Harris J. I., Nature, 203, 30 (1964).
31. Harris J. I., Meriwether P. B., Park J. H., Nature, 198, 154 (1963).
32. Hartley B. S., J. Cell and Compar Physiol., 54, 203 (1959).
33. Hartley B. S., VI Междунар. биохим. конгр., IV Симпоз. Изд-во АН СССР, М. (1962).
34. Hill R. L., Adv. Protein Chem., 37,107 (1965).
35. Hirs C. H. W., J. Biol. Chem., 219, 611 (1956).
36. Hoffmann T., Walsh K- A., Rauffmann D., Neurath H., Fed. Proc., 22, 528 (1963).
37. Holbroock J. J., Pfleiderer G., Schnetger J., Diemair S., Biochem. Z., 344, 1 (1966).
38. Hubbard R. W., Rremer D. M., Anal. Biochem., 12, 593 (1965).
39. Jansen E. F., Curl A. L., Laurence A., Batts A. R., J. Biol. Chem., 179, 189 (1949).
40. Jansz H. S., Brons D., Biochim. Biophys. Acta, 33, 396 (1959).
41. Jansz H. S., Brons D., Warringa M. G., Biochim, Biophys. Acta, 34, 573 (1959).
42. Lai C. Y., Arch. Biochim. Biophys., 128, 22 (1968).
43. Larsen I., Scientific Tools, 12, 24 (1965).
44. Lathe G. H., Ruthaven G. R. J., Biochim. J., 62, 665 (1965).
45. Lawson W. B., Gross E., Folitz C. H., Witkop B., J. Am. Chem. Soc., 85, 1509 (1961).
46. Levy A., Nature, 174, 126 (1954).
47. Li T. R., Vatlee B. L., Biochemistry, 3, 869 (1964).
48. Lindley H., J. Am. Chem. Soc., 77, 4027 (1956).
49. Margoliash E., J, Biol. Chem., 237, 2161 (1961).
50. Michl M., Monatsh. Chemie, 82, 489 (1951).
51. Mikes O., Coll. Czechoslov. Chem. Commun., 22, 831 (1957).
52. Milstein C., Sanger F., Biochim. J., 79, 456 (1961).
53. Monier R., Jutisz M., Bull Soc. Chim. Biol., 32, 228 (1950).
54. Moore S., Bergmann M., J. Biol. Chem., 154, 191 (1944).
55. Moore S., Stein W. H., J. Biol. Chem., 176, 367 (1948).
56. Naughton M. A., Sanger F., Hartley B. S., Shaw D. C., Biochim. J., 77, 149 (1960).
57. Oesterbaan R. A., wan Andrichen M. E., Biochim. Biophys. Acta, 27, 423 (1958).
58. Patchornik A., Lawson W. B., Cross E., Witkop В., J. Am. Chem. Soc., 82, 5923 (1960).
59. Pechere J. F., Dixon G. H., Mayburry R. H., Neurath H., J. Biol. Chem., 233, 1364 (1968).
60. Piez К. A., Morris L., Anal. Biochem., I, 187 (1960).
61. Porath J., Flodin. O., Nature. 183, 1657 (1959).
62. Porter R. R., Sanger F-, Biochem. J., 42, 287 (1948).
63. Raftery M. A., Cole R. D., Biochem. Biophys. Res. Commun., 10, 464 (1963).
64. Ramachandran L. R., Witkop B., J. Am. Chem. Soc., 81, 4028 (1959).
65. Rees M. W., Biochem., J., 40, 632 (1946).
66. Rovery M., Fahre S., Bull. Soc. Chim. Biol., 35, 541 (1953).
67. Rydon H. N., Smith P. W. G., Nature, 169, 922 (1952).
68. Sajgó M., Acta Biochim. Biophys. Acad. Sci. Hung., 4, 385 (1969).
69. Sajgó M., FEBS Lett., 12, 349 (1971).
70. Sanger F., Proc. Chem. Soc., p. 76 (1963).
71. Sanger F., Shaw D. C., Nature, 187, 872 (1960).
72. Sanger F., Tuppy H., Biochem. J., 49, 463; 481 (1951).
73. Schaffer N. K-, Harmann S., Engle R. J., J. Biol. Chem., 214, 799 (1955).
74. Schaffer N. R-, May C. S., Summerson W. H., J. Biol. Chem., 206, 201 (1954).
75. Schwarts J. H., Crestfield A. M., Lipmann T., Proc. Nat. Acad. Sci. (Wash) 49, 722 (1963).
76. Sjöquist J., Biochim. Biophys. Acta, 41, 20 (1960).
77. Smith E. L., Fed. Proc., 16, 801 (1957).
78. Smith E. L., Kimmel J. R., in: Boyer P., Lardy H., Myrbäck К., The Enzymes, Academic Press, New York, 133 (1960).
79. Sosakowa S., J. Biochem. (Tokyo), 53, 188 (1963).
80. Spackman D. H., Fed. Proc. 22, 244 (1963).
81. Spackman D. H., Smith E. L., Brown D. M., J. Biol. Chem., 212, 255 (1955)
82. Synge R. L. M., Chem. Rev., 32, 135 (1943).
83. Synge R. L. M., Tiselius A., Biochem., J., 46, P41 (1950).
84. Tatian H. H., Biochim. Biophys. Acta, 27, 407 (1958).
85. Thompson E. О. P., Biol. Chem., 207, 563 (1954).
86. Toennies G., Homiller R. P., J. Am. Chem. Soc., 64 , 3054 (1942).
87 Tuppy H., Z. Naturforsch., 12b, 784 (1957).
88. Tuppy H., Naturwissenschaften, 46, 35 (1959).
89. Tuppy H., Bodo G., Monatsh. Chemie, 85 , 807, 1024, 1182 (1954).
90. Tuppy H., Dus К-, Monatsh. Chemie. 89. 407 (1958).
91. Tuppy H., Peteus S., Acta Chem. Scand., 9, 363 (1955).
92. Vanecek J., Meloun B., Kostka V., Keil В., Sorm F., Biochim. Biophys. Acta, 37, 169 (1960).
Рекомендуемая литература
Colowick S. P., Kaplan N. O. eds., Methods in Enzymology, vol. XXV, Academic Press, New York, London, 1972.
Dayhoff М., О., Atlas of Protein Sequence and Structure, vol. 5, 1972, Suppl. 1, Natl. Biomed. Res. Foundation Georgetown Univ. Med. Centr. Washington D. C., 1973.
Needleman S. B. ed., Protein Sequence Determination, Springer Verlag, Berlin, Heidelberg, New York, 1970.