Аминокислоты, пептиды и белки - Дэвени Т., Гергей Я. 1976
Ионообменная хроматография в фиксированном слое ионообменника
Области применения
РАЗДЕЛЕНИЕ АРОМАТИЧЕСКИХ И ОСНОВНЫХ АМИНОКИСЛОТ [3]
Ароматические и основные аминокислоты на пластинке “Фиксиоц 50 x 8” разделяются при одномерной хроматографии в цитратном буферном растворе pH 5,23 с концентрацией Na+ 0,35 М (буферный раствор В, табл. 10), который используется в двухколоночной системе аминокислотного анализатора. Типичная хроматография такого разделения представлена на фиг. 49. Колебания pH и концентрации буферного раствора не существенны для фракционирования. Хроматографию проводят при комнатной температуре без предварительного уравновешивания. В камеру наливают слой буферного раствора высотой примерно 1 см. При хроматографии фронт буферного раствора должен подняться на высоту 15 см. Если пластинка не уравновешена, на это уходит около 2 ч. На уравновешенной пластинке (см. буферный раствор для уравновешивания, табл. 10) это происходит за несколько минут. На примере разделения ароматических и основных аминокислот можно оценить высокую разрешающую способность ионообменной хроматографии в тонком слое по сравнению с соответствующей колоночной техникой. Известно, что на малой колонке в этом же буферном растворе (т. е. 0,35 М Na+ pH 5,23) ароматические аминокислоты не отделяются друг от друга.
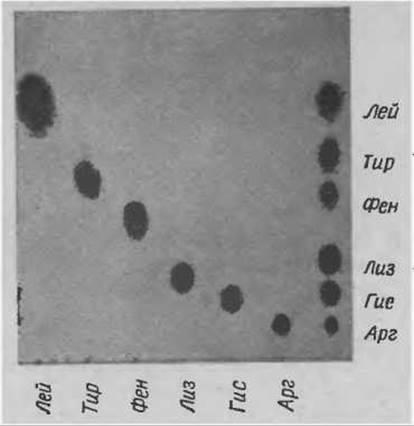
Фиг. 49. Разделение основных и ароматических аминокислот на пластинке “Фиксион 50x8”.
Таблица 10 Приготовление буферных растворов
Буферный раствор для уравновешивания |
А |
Б |
B |
Г |
Д |
Е |
Ж |
|
pH |
3,28 |
3,28 |
3,3 |
5,23 |
4,25 |
6,0 |
4,25 |
6,0 |
Концентрация Na+, М |
0,02 |
0,2 |
0,4 |
0,35 |
0,4 |
1,5 |
0,8 |
1,5 |
Концентрация цитрата, М |
0,0067 |
0,067 |
0,4 |
0,177 |
0,067 |
0,03 |
0,067 |
0,03 |
Лимонная кислота х Н2О, г |
1.4 |
14,1 |
84,0 |
24,6 |
14,1 |
7,0 |
14,1 |
7,0 |
NaOH, г |
0,8 |
8,0 |
16,0 |
14,0 |
8,0 |
4,0 |
8,0 |
4,0 |
NaCl, г |
— |
— |
— |
— |
11,7 |
81,9 |
35,0 |
81,9 |
37%-ная НСl (уд. в. 1,19), мл |
1,2 |
12,3 |
5,9 |
6,5 |
8,4 |
— |
8,4 |
— |
Глицерин, мл.. |
— |
100 |
— |
— |
— |
— |
— |
— |
Метилцеллозольв, мл |
— |
— |
— |
— |
100 |
— |
— |
|
Конечный объем, мл |
1000 |
1000 |
1000 |
1000 |
1000 |
1000 |
1000 |
1000 |
Хорошее разделение можно получить при хроматографии в летучем буферном растворе пиридин — уксусная кислота pH 5, который готовят на деионизованной воде, смешивая 40 мл уксусной кислоты и 40 мл пиридина и доводя конечный объем до 1000 мл. В этом буферном растворе в отличие от цитратного Тир и Фен меняются местами (фиг. 50), а аминокислоты в направлении от линии старта к фронту растворителя движутся в следующем порядке: Apr, Гис, Лиз, Тир, Фен.
В исследованиях, проводимых в сельском хозяйстве и клинической практике, разделение основных и ароматических аминокислот имеет большое значение.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЛИЗИНА ПРИ ПРОВЕДЕНИИ МАССОВЫХ АНАЛИЗОВ В СЕЛЬСКОМ ХОЗЯЙСТВЕ
Определение Лиз необходимо, например при селекции растений, при оценке кормов и т. п. Принцип работы с большим числом проб заключается в следующем [8].
Исследуемые образцы (например, зерна) гидролизуют в течение 40 ч при 105°С в атмосфере азота в 6 н. HCl. Из каждой пробы отбирают порции с одинаковым содержанием азота. Наш опыт показывает, что наилучшие результаты получаются с пробами, содержащими примерно 8—10 мг азота, однако это зависит также от среднего содержания Лиз (или другой аминокислоты, например Мет) в данной пробе. Эквимолярные по азоту порции из каждой пробы наносят непосредственно на пластинку в виде полосы. Если проба содержит, например, 8 мг азота, то при среднем содержании Лиз для удовлетворительной оценки пятен оптимально наносить в односантиметровую полосу 10 мкл раствора.
После нанесения полос гидролизатов параллельно наносят контрольные смеси. Для лучшей идентификации целесообразно наносить на пластинку несколько проб с известным содержанием Лиз. Например, если среднее количество Лиз в 1-сантиметровой полосе 0,5 мкг (исходя из навески и количества нанесенного образца), следует наносить на пластинку 0,25, 0,5 и 0,75 мкг Лиз. При сравнении исследуемых образцов с контролем можно все образцы разделить на 3 типа, в которых содержание Лиз будет низким, средним и высоким. На основе этих данных отбирают те пробы, для которых можно провести количественный анализ. Поскольку содержание всех остальных аминокислот по сравнению с содержанием Лиз овольно велико, рекомендуется хроматографировать пробы по крайней мере до тех пор, пока фронт буферного раствора не поднимется на высоту 17—18 см. Таким же способом можно определить в образцах содержание Мет. Типичная картина разделения Показана на фиг. 50.
Фиг. 50. Определение лизина в гидролизатах растительных белков на пластинке “Фиксион 50x8”.
РАЗДЕЛЕНИЕ АРОМАТИЧЕСКИХ И ОСНОВНЫХ АМИНОКИСЛОТ В КЛИНИЧЕСКОЙ ПРАКТИКЕ
Метод применим для раннего выявления нарушений аминокислотного метаболизма у новорожденных. Эти наследственные дефекты обмена проявляются в накоплении в крови и моче определенных аминокислот. Среди заболеваний, связанных с обменом аминокислот, самым распространенным является фенилкетонурия, при которой в крови накапливается Фен. Кроме того, известны заболевания, характеризующиеся накоплением Лиз, Гис, Тир, а также другими, менее специфическими нарушениями. Так, например, для цистинурии характерно не только повышение количества цистина, но и накопление гомоцистина.
Одно из достоинств тонкослойной ионообменной хроматографии состоит в том, что она является простым и доступным методом раннего обнаружения почти всех наследственных дефектов обмена аминокислот. Нами разработаны две методики для массовых испытаний.
ХРОМАТОГРАФИЯ АРОМАТИЧЕСКИХ И ОСНОВНЫХ АМИНОКИСЛОТ ГЕПАРИНИЗИРОВАННОЙ КРОВИ [3]
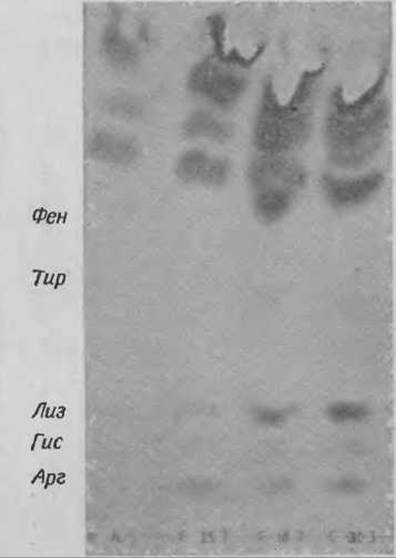
50 мкл крови, взятой с гепарином, вносят в маленькую пробирку и добавляют V, объема трихлоруксусной кислоты (ТХУ). После центрифугирования 20 мкл надосадочной жидкости наносят на пластинку “Фйксион 50 х 8” и дважды хроматографируют вместе с контрольной смесью, нанесенной по обе стороны от опытной пробы. Для удаления ТХУ сначала хроматографируют в 0,01 н. НСl до тех пор, пока фронт буферного раствора не пройдет 18 см. На пластинке с сильным катионообменником ТХУ не связывается и движется вместе с фронтом растворителя, тогда как большинство аминокислот, в первую очередь ароматические и основные, связывается. Пластинку высушивают феном и затем подвергают хроматографии в буферном растворе цитрата натрия pH 5,23 (буфер В, табл. 10). Типичная хроматограмма представлена на фиг. 51.
Фиг. 51. Хроматограмма надосадочной жидкости плазмы крови после обработки трихлоруксусной кислотой.
К — контрольная смесь аминокислот (с последовательно возрастающими Rf) Apr, Гис, Лиз, Фен, Тир, Лей. 1, 2 — пробы из плазмы больного фенилкетонурией; 3, 4, 5 — пробы из плазмы здорового человека.
ОПРЕДЕЛЕНИЕ СВОБОДНЫХ АМИНОКИСЛОТ В ПРОБЕ КРОВИ, ВЫСУШЕННОЙ НА ФИЛЬТРОВАЛЬНОЙ БУМАГЕ [7]
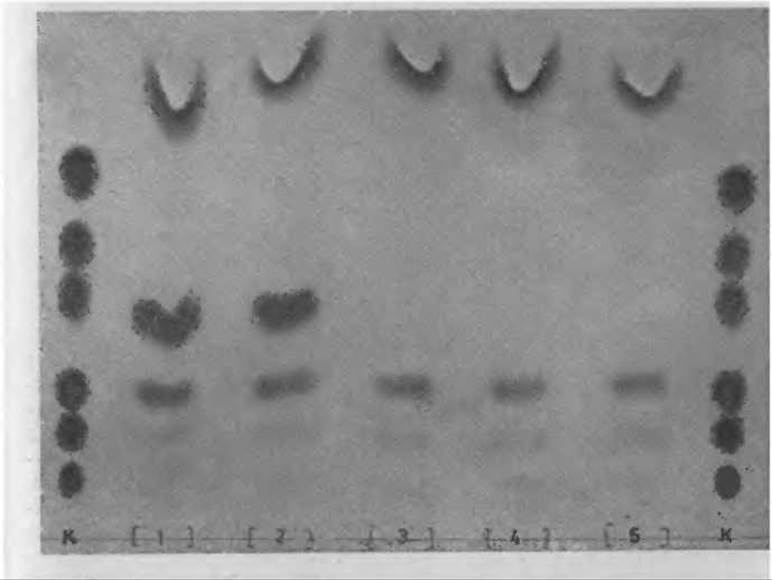
Одну каплю крови (примерно 50 мкл) наносят на тонкую фильтровальную бумагу и дают ей высохнуть при комнатной температуре; диаметр пятна крови должен быть около 15 мм. С помощью пробойника вырезают из пятна 5—6 дисков диаметром около 5 мм и помещают их в вассермановскую пробирку, куда затем наливают 0,1 мл раствора 0,1 н. НСl в 95%-ном этаноле. Пробирки закрывают парафильмом и оставляют на ночь при комнатной температуре. На следующий день образцы наносят на пластинки “Фиксион 50 х 8” по 10—15 мкл в одну точку. Благодаря присутствию в растворе спирта с помощью фена удается наносить образцы довольно быстро. После нанесения проводят хроматографию в нитратном (или каком-либо другом) буферном растворе pH 5,23. Типичная картина такого фракционирования показана на фиг. 52.
Этот способ очень удобен для анализа проб крови: кровь, взятую из пальца или из пятки, после непосредственного нанесения на фильтровальную бумагу можно отправлять по почте. Кислотноспиртовое элюирование в. данном случае позволяет обойтись без предварительного удаления белковых веществ, а также повторной хроматографии. При более низком значении pH и более высокой концентрации Na+ основные компоненты разделяются на большее число фракций: Ори, например, можно отделить от Лиз [10]. Для этой цели применяют буферный раствор Г (табл. 10) с концентрацией Na+ 0,4 М и pH 4,25.
Фиг. 52. Исследование образцов крови, высушенных на фильтровальной бумаге, при диагностике фенилкетонурии.
К — контрольная смесь аминокислот Apr, Гис, Лиз, Фен, Тир, Лей, 1, 4 — кровь больного фенилкетонурией; 2, 3, 5 — кровь здорового человека.
ОДНОМЕРНОЕ РАЗДЕЛЕНИЕ АМИНОКИСЛОТ [4]
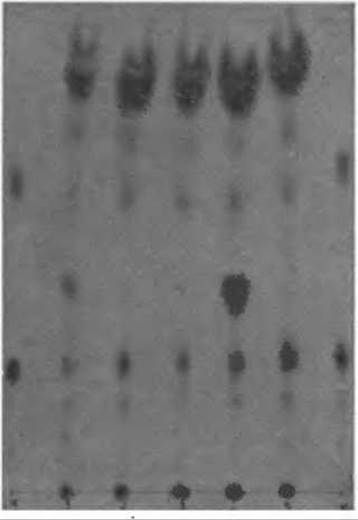
С помощью тонкослойной ионообменной хроматографии можно разделить-16 аминокислот, присутствующих в белковом гидролизате, используя всего лишь один буферный раствор. В буферном растворе Б (табл. 10), имеющем относительно высокую концентрацию ионов цитрата, смесь из 16 аминокислот делится на 15 компонентов. Картина такого разделения показана на фиг. 53. Из 16 аминокислот не разделяются только Тре и Сер.
Около 0,5 мкг исследуемого и контрольного (смесь аминокислот или отдельные аминокислоты) материала наносят в виде точки на уравновешенную пластинку. Тщательно вымытую хроматографическую камеру заполняют буферным раствором до высоты 1 см, закрывают ее и помещают в воздушный термостат (45°С) на 15 мин. Нижний край пластинки погружают в подогретый буферный раствор и проводят хроматографию, позволяя ему подниматься до высоты 18 см (около 3,5 ч). После высушивания хроматограмму проявляют коллидин-нингидриновым реагентом. Фракционирование очень чувствительно к колебаниям pH и температуры. Повышение pH на 0,1—0,2 единицы влечет за собой резкое увеличение Rf, что может улучшить разделение в нижней части хроматограммы — где мигрируют основные и ароматические аминокислоты (Лей, Иле, Мет и Вал), однако, разделение в верхней части хроматограммы — где мигрируют аминокислоты от Ала до Асп, ухудшается
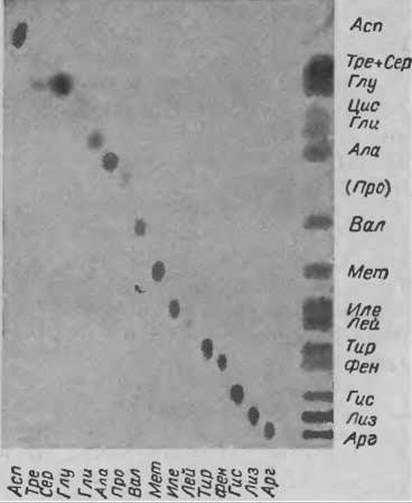
Фиг. 53. Разделение аминокислот на пластинке “Фиксион 5Gх8”.
Одномерная тонкослойная ионообменная хроматограмма дает почти те же значения Rf аминокислот, что и диаграмма, полученная на аминокислотном анализаторе. Разделение аминокислот от Асп до Ала в анализаторе также чувствительно к изменению pH и молярности буферного раствора и ухудшается при увеличении pH. В то же время разделение аминокислот от Вал и далее уже нечувствительно к изменению pH и молярности.
Следовательно, если, например при хроматографии в тонком слое, основные и нейтральные аминокислоты до Вал имеют необычно высокие значения Rf и вместе с тем разделение в верхней части пластинки оказалось неполным, можно сделать вывод, что pH буферного раствора выше 3,3. К сожалению, большинство приборов для измерения pH недостаточно надежны и точны, поэтому приходится поступать так, как это принято при фракционировании в анализаторе, где pH буферного раствора проверяют по “месту цистина”. В тонкослойной хроматографии pH также проверяют по относительной подвижности компонентов. В оптимальном варианте цистин располагается между Ала и Вал.
Если значение Rf Вал близко к 0,5, а Асп — к 0,9, но последняя еще не уходит с фронтом растворителя, то pH буфера оптимален. Колебания температуры в первую очередь отражаются на разделении Мет, Иле и Лей. При 45—509С разделение этих трех аминокислот удовлетворительно и вполне воспроизводимо. При понижении температуры разделение ухудшается, а при температуре ниже 40°С аминокислоты вообще не разделяются. По-видимому, температура 45°С является оптимальной, так как, с одной стороны, достигается полное разделение этих трех аминокислот, а с другой — при такой температуре еще нет необходимости в добавлении специальных веществ (например, глицерина), уменьшающих испарение буфера, которые могут влиять на разделение в верхней части хроматограммы.
Следует подчеркнуть, что ионообменная хроматография в тонком слое не годится для количественного определения аминокислотного состава белковых гидролизатов, так как она дает только качественную картину. Зато этот метод вполне применим для исследования гидролизатов пептидов и пептидных остатков при расшифровке последовательности аминокислот. Он позволяет определить молярные пропорции обнаруженных аминокислот — обычно по отношению к какой-либо данной аминокислоте. Если, например, исследуют трипсиновый гидролизат, то таким эталоном для сравнения служит Лиз или Apr. Как правило, оценку производят визуально или на денситометре. Этим методом можно определить соотношение компонентов в пределах 4—5-кратных различий (например, молярные соотношения в пептиде Лиз1Вал2Глу2Тре3). При меньших различиях относительных молярных концентраций аминокислот метод теряет чувствительность. Это не позволяет его использовать для анализа белков, в которых эти различия могут быть очень малы (например, Лиз10Асп55Вал48Глу49 ... и т. д.).
ИОНООБМЕННАЯ ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ ФЕРМЕНТАТИВНЫХ ГИДРОЛИЗАТОВ [11, 12]
Ферментативный гидролиз можно проводить различными способами, один из которых состоит в следующем: в пластмассовой пластинке толщиной 10—15 мм, длиной 10 см и шириной 5 см высверливается 2 ряда углублений. Объем всех, кроме одного, углублений должен быть примерно 50 мкл, а объем крайнего верхнего — в 3 раза больше. Это углубление выполняет роль реакционного сосуда; обозначим его через Р, а все остальные цифрами 1, 2, 3...n. Исследуемый пептид (около 0,05 мкмоль) в 0,1 М бикарбонате аммония вносят пипеткой в углубление Р. Во все остальные углубления наливают по 10 мкл 1 н. НСl. После этого в углубление Р добавляют соответствующий фермент (например, карбоксипептидазу А, карбоксипептидазу В или их смесь, а в случае N-концевого анализа — аминопептидазу). Количество фермента составляет 1/20 часть количества пептида. Сразу после добавления фермента 10 мкл смеси из “реакционного сосуда” переносят в углубление 1, где уже находится НСl.
Пластинку покрывают парафильмом (или стеклянной пластинкой) и ставят в воздушный термостат при 37°С. Через определенные промежутки времени (целесообразно через 30, 60, 90, 120 мин) отбирают пробы из углубления Р и переносят в углубления 2, 3, 4... и т.д. Таким образом можно проследить за кинетикой гидролиза, происходящего в углублении Р. По окончании реакции содержимое углублений переносят на пластинку “Фиксион 50 х 8”, на которую наносят также и контрольную смесь. Хроматографию проводят в буферном растворе pH 3,3 при 45°С. На проявленной пластинке фактически представлена кинетика гидролиза. Материал, взятый в начале гидролиза (нулевая точка — углубление 1), сравнивают с гидролизатами, взятыми через 30 (углубление 2), 60 (углубление 3), 90 (углубление 4) и 120 мин (углубление 5). Практически этим микрометодом за 2 ч можно достоверно определить порядок-3—5 аминокислотных остатков в 8—10-членном пептиде. Условия эксперимента — соотношение фермента и субстрата, время инкубации, моменты отбора проб — можно варьировать в зависимости от природы субстрата, а также от цели анализа.
Количественные определения проводят способами, используемыми в обычной тонкослойной хроматографии. Хроматограммы на пластинках “Фиксион” денситометрируют точно так же, как и в случае других носителей (целлюлоза, силикагель). Если по какой-то причине (отсутствие приборов, нестабильность цветной реакции и т. п.) это неосуществимо, тогда соответствующие зоны можно элюировать. В связи с тем, что катионообменная смола обладает большой сорбцией, необходимо обращать внимание на то, чтобы элюирующий раствор имел pH 6 или выше. Если полуколичественные методы, применяемые в обычной тонкослойной хроматографии, дают неудовлетворительные результаты, нужно воспользоваться аналогичным количественным методом с использованием анализатора. Для определения полного аминокислотного состава целесообразно применять одноколоночный двухбуферный [1] или трехбуферный [2] метод (см. гл. VI).
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ МЕТИОНИНА И ЛИЗИНА В ОБРАЗЦАХ РАСТИТЕЛЬНОГО И ЖИВОТНОГО ПРОИСХОЖДЕНИЯ [8]
В работах по селекции растений и оценке кормов знание количества Лиз и Мет особенно важно. Совсем не обязательно при этом проводить количественный анализ огромного числа образцов. Предварительный отбор на основе приближенных данных дает вполне достаточную информацию о том, в каких именно образцах следует провести полный количественный анализ.
Для определения Мет в природных веществах можно использовать гидролизаты, полученные описанным выше методом. Они, как правило, содержат значительно большие количества Лиз, чем Мет. В связи с этим в образце с низким содержанием Мет довольно трудно на одной пластинке провести количественное определение этих двух аминокислот. Целесообразно проводить анализ Лиз и Мет в одном гидролизате на двух пластинках.
Гидролиз проводят по ранее описанному методу. Не удаляя кислоты, 20 мкл гидролизата в виде полосы шириной 1 см (если проба содержит 8 мг азота в 2 мл 6 н. НСl) наносят на уравновешенную пластинку “Фиксион 50 х 8” и подсушивают феном. Хроматографию проводят в буферном растворе А (табл. 10), который является первым буферным раствором анализатора, но дополнительно содержит 10% глицерина. Рекомендуется хроматографировать при 45°С и дать возможность фронту раствора подняться до верхнего края пластинки. Если содержание Мет в образце очень мало, то пластинку следует постепенно погружать в растворитель все глубже и проводить проточную хроматографию с помощью фильтровальной бумаги, как это делается при уравновешивании. При этом компоненты с высокими элюируются с пластинки, что облегчает отделение Мет от Вал, Лей и Иле, которых, вероятно, значительно больше в смеси, чем Мет. Хроматограмму проявляют кадмий-нингидрином при 45 С или при комнатной температуре. Картина хроматографического разделения представлена на фиг. 54.
Для количественной оценки применяют любой из способов обычной тонкослойной хроматографии: денситометрию, элюирование и т. п. Если требуется определить содержание Лиз и Мет очень точно, можно использовать короткие экспресспрограммы аминокислотного анализатора [3] (см. гл. VI).
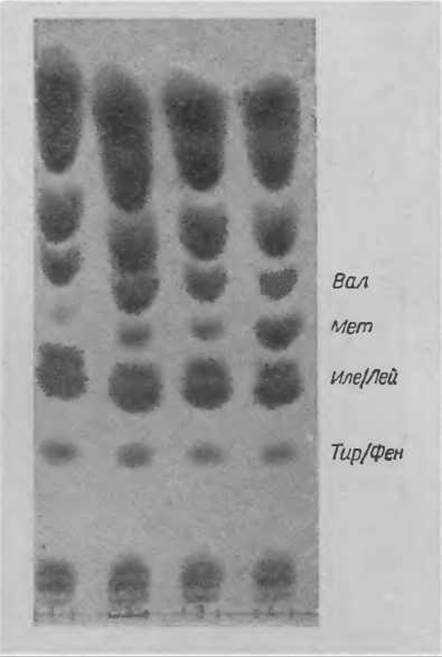
Фиг. 54. Определение метионина в гидролизатах растительных белков.
1 — низкое содержание Мет; 2,3 — среднее содержание Мет; 4 — высокое содержание Мет.
ОПРЕДЕЛЕНИЕ ТРИПТОФАНА В ЩЕЛОЧНОМ ГИДРОЛИЗАТЕ [5]
В кислотном гидролизате Три определить нельзя, так как при этом он полностью разлагается. В принципе существуют 2 методики определения Три: специфическая качественная цветная реакция в щелочном гидролизате и спектрофотометрия Три в исходном растворе. Обе методики имеют свои недостатки: цветная реакция в щелочном гидролизате не строго специфична, так как гидролизат содержит много примесей, а спектрофотометрический способ применим только тогда, когда вещество полностью растворяется, образуя прозрачный раствор, или смесь не содержит компонентов (например, коферментов), поглощающих при данной длине волны. Методы классической хроматографии в тонком слое или на бумаге не позволяют достоверно определить Три из-за высокой концентрации солей в нейтрализованном гидролизате. При тонкослойной ионообменной хроматографии удается преодолеть это затруднение, так как высокое содержание соли не мешает разделению.
ГИДРОЛИЗ
Гидролиз проводят следующим образом: примерно 5 мг пептида или белка растворяют в ампуле в 2 мл 2 н. NaOH, заполняют ампулу азотом и запаивают ее. Гидролиз проводят в течение 5 ч при 105°С. После охлаждения добавляют 3 мл буферного раствора А и 1 мл концентрированной НСl.
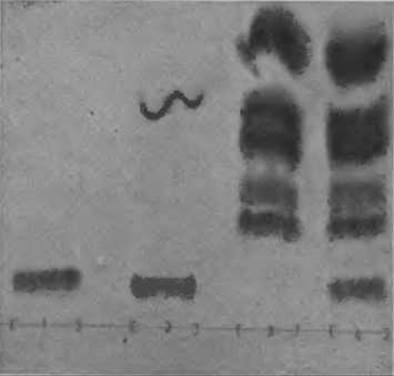
Фиг. 55. Определение триптофана на пластинке “Фиксион 50 х 8”.
1 — ацетилтриптофаи; 2 — гидролизат глицилтриптофана; 3 — гидролизат пептида, не содержащего Три; 4 — гидролизат пептида, содержащего Три.
ХРОМАТОГРАФИЯ
Для разделения используют буферный раствор Д (табл. 10), содержащий 10% метилцеллозольва (третий буферный раствор одноколоночной системы). Типичная хроматограмма показана на фиг. 55.
При относительно высоком pH [6] и концентрации Na+ 1,5 М Три имеет самое низкое значение При хроматографии щелочного гидролизата пептидов для получения четкой картины достаточно продвижения фронта растворителя на высоту 8—10 см. Если требуется количественно определить Три, можно прибегнуть к методике, разработанной для анализатора (экспресспрограмма):
Высота смолы в колонке |
14 см |
Буферный раствор |
Д (табл. 10) |
Скорость подачи буферного раствора |
100 мл/ч |
Скорость подачи нингидрина |
50 мл/ч |
Температура |
55°С |
Давление буферного раствора |
8—12 атм |
ОПРЕДЕЛЕНИЕ РАЦЕМИЗАЦИИ В ПЕПТИДНОМ СИНТЕЗЕ [3]
Иногда в пептидном синтезе требуется провести количественную оценку рацемизации, происходящей по мере протекания реакции. Это имеет особое значение в многоступенчатых сложных синтезах, когда превращение определенных промежуточных продуктов является критическим этапом образования конечного продукта. Для обнаружения рацемизации японскими авторами была разработана остроумная методика, которую можно широко использовать на различных стадиях пептидного синтеза. Ее суть заключается в следующем. В соответствующих условиях реакции в процессе синтеза происходит взаимодействие дипептида Гли-L-Ала с L-Лей. В оптимальном варианте продуктом реакции является исключительно Гли-L-Ала-L-Лей. Если же происходит рацемизация, то наряду с этим продуктом в реакционной смеси появится трипептид Гли-D-Ала-L-Лей. Для обнаружения и определения содержания этих двух трипептидов японские авторы использовали аминокислотный анализатор. Правда, разработанная ими методика не подходит для анализа большого количества проб, так как с учетом времени регенерации на исследование одного образца требуется примерно 3,5 ч.
На пластинке “Фиксион 50x8” изомеры-трипептиды Гли-L-Ала-L-Лей и Гли-D-Ала-L-Лей разделяются довольно хорошо. По интенсивности пятен разделенных пептидов можно оценить степень рацемизации препарата. Хроматографию проводят на Уравновешенных пластинках в буферном растворе Е (табл. 10), т. е. во втором буферном растворе одноколоночной системы анализатора. Для получения оптимального разделения на пластинку наносят 10 мкл раствора образца в 0,01 н. НСl (концентрация пептида 10 мг/мл). Хроматографируют при комнатной температуре до высоты 12—15 см. Если требуется количественно определить степень рацемизации (с точностью 5—10%), применяют денситометрию тонкослойных хроматограмм. Для получения более точных результатов целесообразно воспользоваться автоматическим анализатором.