Основы биохимии - А. А. Анисимов 1986
Ферменты (энзимы)
Кинетика ферментативных реакций
3.7.1. Единицы ферментативной активности. Кинетика — это учение о скоростях реакций, их зависимости от различных факторов. Скорость ферментативной реакции, как и любой химической реакции, определяется количеством веществ, прореагировавших за единицу времени при заданных условиях. Скорость ферментативной реакции зависит от активности фермента, которая может быть выражена в различных единицах. До недавнего времени наиболее принятой единицей активности была так называемая стандартная единица (Е). Под ней понимают то количество фермента, которое катализирует превращение 1 мкМ субстрата в 1 мин при оптимальных условиях для данного фермента (t°, pH, [S]). Согласно последнему международному соглашению 1 единица любого фермента есть такое количество фермента, которое в определенных условиях катализирует превращение субстрата со скоростью 1 моль/с. Эта единица называется катал (сокращенно кат; 1 кат = 6⋅107 стандартных единиц). Для выражения активности фермента обычно используют нано- и пикокаталы.
Существует также понятиe удельной активности, которая выражается числом единиц активности фермента, приходящихся на 1 мг белка в ферментативном препарате (Е/мг). Удельную активность в настоящее время рекомендуют выражать в кат/кг. Если известна молекулярная масса фермента, то можно рассчитать молекулярную активность (число оборотов), она характеризуется числом молей субстрата, которое подвергается превращению 1 молем фермента за 1 мин. В том случае, когда у фермента несколько активных центров, то определяют число молей субстрата, подвергающихся превращению одним молем каталитических центров (молярная концентрация фермента, умноженная на число активных центров в молекуле фермента) — активность каталитического центра.
При определении активности ферментов измеряют начальные скорости реакции (V) в стационарной фазе (рис. 3.10). Участок реакции, предшествующий установлению стационарного состояния, называют переходным. После стационарной фазы скорость реакций снижается.
Это может быть связано с уменьшением концентрации исходного субстрата, накоплением продуктов реакции и ингибированием ими фермента, инактивацией фермента в процессе инкубации и рядом других причин.
3.7.2. Влияние концентрации фермента и субстрата на начальную скорость реакции. Зависимость скорости реакции от концентрации фермента является линейной (рис. 3.11). Нелинейная зависимость при высоких концентрациях фермента наблюдается вследствие нехватки субстрата или активатора, агрегации молекул ферментативного белка, приводящей к маскировке активных центров. Отсутствие прямо пропорциональной зависимости при небольших концентрациях фермента может быть результатом присутствия в инкубационной среде токсических примесей, связывающихся с ферментом и инактивирующих его.

Рис. 3.10. Зависимость скорости ферментативной реакции от времени:
I — переходный участок. II — участок начальной скорости, III — участок основного протекания реакции
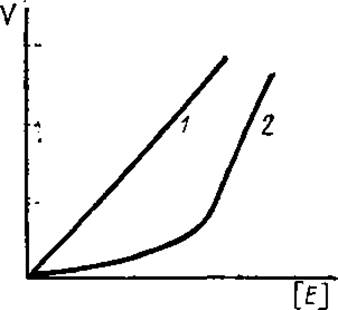
Рис. 3.11. График зависимости начальной скорости реакции (V) от концентрации фермента [F]:
1 — норма, 2 — в системе присутствует небольшое количество токсических для фермента примесей

Рис. 3.12. Зависимость скорости реакции (V) от концентрации субстрата [S]
Почти во всех случаях график зависимости начальной скорости реакции от концентрации субстрата представляет собой гиперболу (рис. 3.12).
По мере возрастания концентрации субстрата кривая выходит на плато, скорость достигает своей максимальной величины (Vmах). Это говорит о том, что все молекулы фермента (их активные центры) полностью насыщены субстратом.
Гиперболический график зависимости V от [S] показывает, что порядок реакции при ферментативном катализе изменяется. При небольших концентрациях субстрата протекает реакция 1-го порядка (V пропорционально [S]). При насыщающей концентрации субстрата скорость не зависит от нее, такая реакция является реакцией нулевого порядка. При промежуточных концентрациях субстрата протекает реакция смешанного порядка.
Для описания зависимости начальной скорости реакции от концентрации субстрата Л. Михаэлисом и М. Ментен выведено уравнение для односубстратной реакции:
![]()
Это уравнение получило название уравнения Михаэлиса—Ментен (KS — константа диссоциации фермент-субстратного комплекса, равная k-1/k1, Vmax — максимальная скорость реакции). При выводе уравнения Л. Михаэлис и М. Ментен исходили из следующего обобщающего механизма односубстратной реакции. Фермент взаимодействует с субстратом, образуя фермент-субстратный комплекс (ES), который затем распадается на свободный фермент и продукт реакции:
![]()
где k1, k-1, k2, k-2 — константы скоростей соответствующих стадий реакции.
Стадию обратной реакции — образование ES из E и Р — обычно не учитывают, так как рассматривают начальные скорости реакций, при которых только очень небольшая доля S превращена в Р. В этом случае из Р практически не образуется ES.
Позже Дж. Бриггсом и Дж. Холдейном было предложено уравнение Михаэлиса-Ментен записывать в следующем виде:
![]()
где Кm — константа Михаэлиса. Кm = (k-1 + k2)/k1 = KS + k2/k1.
Такое преобразование уравнения связано с тем, что Михаэлис и Ментен исходили из допущения, согласно которому 1-я стадия реакции (E + S = ES) является равновесной, т, е. равновесие в ней достигается гораздо быстрее, чем успевает распасться комплекс ES с образованием продукта реакции. Однако это далеко не всегда справедливо, и хотя указанная стадия протекает достаточно быстро и обратимо; она не является равновесной. Поскольку ферментам свойственна высокая каталитическая активность, в реальных условиях система находится в стационарном состоянии, при котором концентрация промежуточного соединения (ES) постоянна, т. е. скорости его образования и распада, в том числе и до конечного продукта, равны. Это и учитывается введением Кm вместо Ks. Кm ≂ Кs только при k2 ≪ k-1.
Константа Михаэлиса (Km) — важная характеристика фермента, по ней судят о степени сродства фермента к субстрату и способности фермента образовывать продукты реакции. Однако об истинном сродстве фермента к субстрату можно судить только по величине Ks, определить которую экспериментально значительно сложнее, чем Km. Kmизмеряется в моль/л. Если Km велика, то комплекс ESлегко распадается на исходные вещества и реакция идет медленно. При низких Km(велика k+1) реакция идет быстро. Для большинства ферментативных реакций с участием одного субстрата Kmравняется от 1 ⋅ 10-2 до 1 ⋅ 10-5 моль/л. Kmи Vmaxдля данного фермента и субстрата при определенных параметрах внешней среды (pH, t° и др.) —величины постоянные. У ферментов с относительной специфичностью каждый субстрат характеризуется своим значением Кm.
Кm определяют графически по результатам измерения V при различных концентрациях субстрата. В частном случае, когда
![]()
отсюда Km— [S], т. e. константа Михаэлиса равна концентрации субстрата при скорости реакции, равной половине от максимальной скорости (см. рис. 3.12). По гиперболической зависимости V от [S] точное определение Кm затруднено, поскольку Vmах является величиной асимптотической и определяется недостаточно точно. В связи с этим предложено несколько преобразований уравнения Михаэлиса—Ментен, сводящихся к получению прямо пропорциональной зависимости V от [S]. Приравнивая обратные выражения правой и левой частей уравнения Михаэлиса—Ментен, Г. Лайнуивер и Д. Бэрк получили следующую зависимость:
![]()
где Km, Vmах — величины постоянные при заданных условиях определения, а К и [S] — переменные. Это уравнение прямой линии (y = kx+b). График зависимости 1/V от 1/[S] представляет собой прямую с наклоном Km/Vmax, отсекающую на оси ординат отрезок 1/Vmах, а на оси абсцисс — 1 /Кm (рис. 3.13). Измеряя отрезок на оси абсцисс, определяют величину Кm.
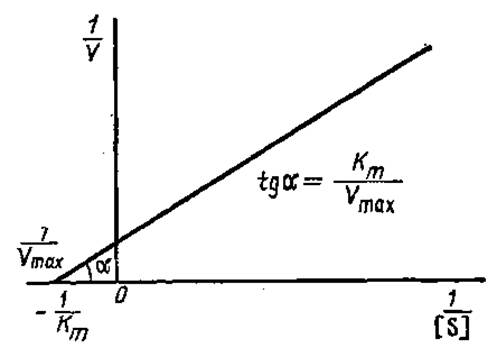
Рис. 3.13. График Лайнуивера—Бэрка
При умножении обеих частей уравнения Лайнуивера—Бэрка на [S], получают новое линейное уравнение, которое также используют для графического определения Кm (рис. 3.14):
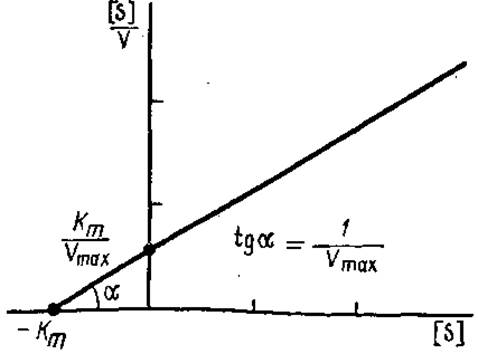
Рис. 3.14. График зависимости [S]/V от [5], используемый для определения Кm и Vmах

Рис. 3.15. Зависимость V от V/[S] (график Эди-Хофсти)
![]()
Умножение обеих частей уравнения Лайнуивера—Бэрка на Vmах дает еще один вид уравнения, график которого представляет собой прямую линию (рис. 3.15):
![]()
Э. Корниш-Боуденом предложен следующий линейный график для определения Кm и Vmax:
![]()
Для построения этого графика не требуется расчетов, каждая прямая на нем соответствует одному наблюдению (рис. 3,16).
При выводе уравнения зависимости скорости реакции от концентрации субстрата Л. Михаэлис и М. Ментен исходили из упрощенной схемы ферментативной реакции. Однако большинство ферментативных реакций не являются односубстратными и, кроме того, в них образуется не один, а несколько фермент-субстратных комплексов: E + S ⇄ ES ⇄ ESP' ⇄ EP ⇄ E + P. Появление дополнительных комплексов не изменяет общего вида уравнения Михаэлиса—Ментен, но Кm и Vmax будут боле сложным константами, включающими в себя большее число констант скоростей, чем в случае одностадийной реакции.
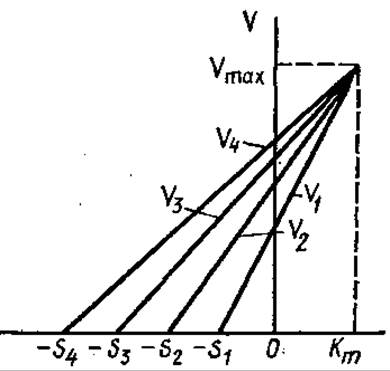
Рис. 3.16. Прямая линейная зависимость Vmax от Кm (график Корниш-Боудена)
Уравнение скорости для двухсубстратных реакций также близко к уравнению Михаэлиса—Ментен: протекание реакций сопровождается образованием фермент-субстратных комплексов, причем каждый субстрат характеризуется своей Кm Константа Михаэлиса определяется, как и в случае односубстратных реакций, графически по зависимости V от концентрации одного из субстратов при постоянной, насыщающей концентрации другого; Кm можно определить и по отношению к активатору фермента, при этом концентрация субстрата должна быть фиксированной.
Построение кинетических кривых зависимости V от [S] оказывается полезным не только при определении величины Кm, но и при установлении механизма протекания двухсубстратных реакций, типа ингибирования (см. разд. 3.7.5).
3.7.3. Влияние температуры на скорость ферментативной реакции. Скорость ферментативной реакции существенно зависит от температуры. Зависимость константы скорости реакции (k) от температуры (Т) выражается уравнением Аррениуса: 2,3 lgk = B—Ea/RT, где Еа — энергия активации (Дж/моль), В — характеризует частоту столкновения молекул и их взаимную ориентацию, Т — абсолютная температура, R — газовая постоянная. В уравнении Еа, В, R — величины постоянные при заданных условиях. Следовательно, график зависимости lgk от 1/T должен быть прямолинейным (рис. 3.17).
Наклон прямой равен Ea/2,3R, поэтому по графику зависимости скорости реакции от температуры можно определить энергию активации реакции. Точнее говоря, из графика определяется только Еа лимитирующей стадии реакции, самой медленной стадии, т. е. стадии с самой высокой энергией активации (рис. 3.18).
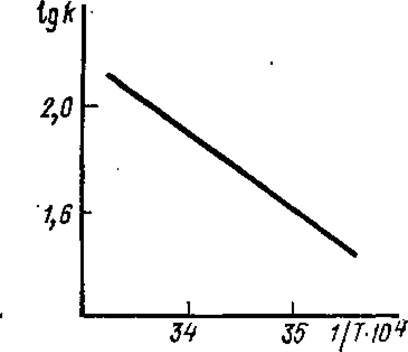
Рис. 3.17. Влияние температуры на константу скорости (k) ферментативной реакции (зависимость lg k от обратной величины абсолютной температуры)
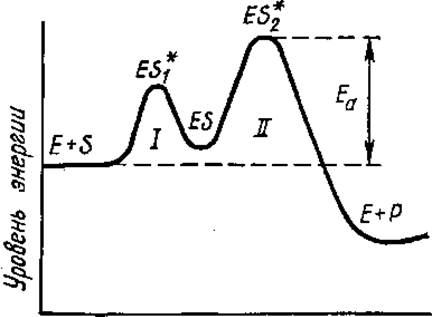
Рис. 3.18. Энергетическая схема ферментативной реакции:
ES* — переходное состояние (активированный комплекс — короткоживущее соединение Е и S, возникающее при их сближении и переходящее затем в обычный комплекс ES — промежуточное состояние); Еа — энергия активации, Р — продукт реакции; I, II — стадии реакции (стадия II — лимитирующая)
График зависимости lgk от 1/T прямолинеен только в определенных пределах. Это связано с тем, что при повышении температуры скорость ферментативных реакций увеличивается до определенного максимального значения, при дальнейшем повышении температуры происходит снижение скорости реакции вследствие начинающейся тепловой денатурации фермента (рис. 3.19). При нагревании выше 80°С преобладающее большинство ферментов полностью денатурирует.
Температурный оптимум для большинства ферментов лежит в пределах 40—60°С. Высокой термостабильностью обладают ферменты термофильных микроорганизмов; некоторые из них способны даже выдерживать кратковременное кипячение без существенного снижения активности. Термостабильность ферментов повышается в присутствии субстрата (ов). В кристаллическом виде ферменты более термоустойчивы. Большинство ферментов хорошо сохраняет свою активность при пониженных и отрицательных температурах. Только некоторые холодлабильные ферменты инактивируются при понижении температуры от 30 до 0°С.
3.7.4. Влияние pH на скорость ферментативной реакции. Графическая зависимость скорости большинства ферментативных реакций от pH имеет колоколообразную форму (рис. 3.20). То значение pH, при котором скорость реакции максимальна, называется оптимумом pH; при отклонении pH в любую сторону от этого значения скорость реакции снижается. Ферментативные реакции чувствительны к изменению pH, потому что ферменты содержат большое число ионогенных групп. Состояние ионизации наиболее важных для функционирования фермента групп, зависящее от pH, влияет на каталитическую активность фермента, поскольку максимальную активность фермент может проявлять только при определенном состоянии этих групп: ионизированном или неионизированном. Во многих случаях к ионизации способны и определенные группы субстрата, что также сказывается на оптимуме pH для действия фермента, так как для реакции важно присутствие субстрата в определенной форме.
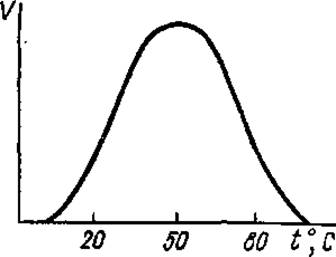
Рис. 3.19. Зависимость скорости ферментативной реакции от температуры

Рис. 3.20. Влияние активной реакции среды на скорость ферментативной реакции
Кроме того, pH может оказывать косвенное влияние на активность ферментов, дестабилизируя их структуру, нарушать прочность связи апофермента с небелковым компонентом. Изменение pH может также влиять на состояние активаторов и ингибиторов ферментов. Значение pH, соответствующее оптимальному, не всегда совпадает со значением pH, характерным для внутриклеточной среды. Поэтому pH может быть одним из факторов, ответственных за регулирование ферментативной активности внутри клетки.
3.7.5. Ингибиторы ферментов. Скорость ферментативной реакции может быть снижена, а в ряде случаев и полностью приостановлена при действии ингибиторов — веществ, которые по той или иной причине частично или полностью препятствуют образованию продуктивного фермент-субстратного комплекса. В частности, токсичность многих ядов для живых организмов, лечебный эффект ряда лекарственных препаратов обусловлены их ингибирующим действием на ферменты. Среди ингибиторов есть как синтетические
вещества, так и природные метаболиты. Для ингибиторов характерна специфичность действия. Необратимую (и неспецифичную) денатурацию ферментов под влиянием тепла, сильных кислот или каких-либо других факторов обычно не рассматривают как ингибирование. К ингибиторам неприродного происхождения относят, например, фторид натрия — ингибитор фосфатаз, фенантролин — металлсодержащих ферментов, р-хлормеркурибензоат — SH-ферментов, диизопропилфторфосфат (ДИФФ) — сериновых протеиназ и эстераз. Действие таких ингибиторов обычно необратимо.
Примерами обратимых ингибиторов являются клеточные метаболиты и их структурные аналоги, снижающие активность ферментов. Различают три типа обратимого ингибирования ферментов: конкурентное, неконкурентное и бесконкурентное. К конкурентным ингибиторам относят вещества, способные связываться с активным центром фермента и, следовательно, конкурировать с субстратом. Взаимодействие фермента с конкурентным ингибитором записывают в виде уравнения: Е + I ⇄ ЕI, где I — ингибитор; El — фермент-ингибиторный комплекс, не превращающийся в продукт; Ki — константа ингибирования, представляющая собой константу диссоциации EI, Кі = [Е][І]/[ЕІ]. Ki показывает сродство ингибитора к ферменту; чем меньше Ки тем сильнее ингибитор.
При Ki → О ингибирование необратимо. Скорость реакции в присутствии конкурентного ингибитора зависит от соотношения [S] и [I]. Примером конкурентного ингибирования является действие малоновой кислоты и ряда других дикарбоновых кислот на сукцинат-дегидрогеназу (СДГ). В качестве конкурентных ингибиторов выступают вещества, структурно сходные с субстратом. В случае СДГ это вещества следующего строения:
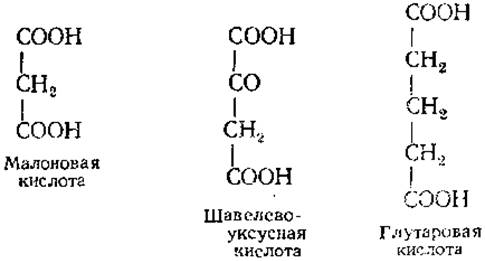
Наиболее сильным ингибитором СДГ из них является малоновая кислота, замедляющая на 50% скорость ферментативной реакции при [I]/[E] = 50. Увеличение концентрации субстрата приводит к снижению степени ингибирования, поскольку субстрат вытесняет ингибитор из активного центра.
Неконкурентный ингибитор связывается с ферментом вне активного центра, поэтому конкурентных отношений между субстратом и ингибитором нет, и степень ингибирования зависит только от концентрации последнего. Связываясь с ферментом, неконкурентные ингибиторы вызывают существенные изменения конформации фермента, пространственной структуры активного центра, что и нарушает нормальное связывание субстрата.
Бесконкурентное ингибирование наблюдается в том случае, когда ингибитор обратимо взаимодействует с ферментом, только после образования ES : ES + I ⇄ ESІ. Бесконкурентное ингибирование наиболее характерно для ферментов, каталитическое действие которых осуществляется по механизму «пинг-понга».
Помимо указанных типов обратимого ингибирования выделяют смешанный тип ингибирования.
Тип ингибирования может быть выявлен с помощью кинетического анализа, при изучении зависимости V от [S] при различных концентрациях ингибитора (рис. 3.21). Влияние конкурентного ингибитора на скорость реакции заключается в том, что он увеличивает Km и оставляет без изменения Vmах. Для бесконкурентного ингибирования характерно уменьшение в одинаковой степени Vmах и Kmи вследствие этого постоянство отношения Vmах/Km.
Неконкурентные ингибиторы действуют на активность фермента, снижая значение Vmах и не оказывая влияния на Кm.
В случае построения зависимости Vmах от Кm тип ингибирования характеризуется смещением точки пересечения. Для конкурентного — вправо, бесконкурентного — по направлению к началу координат, для смешанного характерен промежуточный характер смещения.
Многие фармакологически активные соединения изменяют течение метаболических процессов в результате действия на ферменты. Некоторые из них выступают в роли конкурентных ингибиторов благодаря сходству структуры с метаболитами. Такие соединения называются антиметаболитами. Например, синтетические антибактериальные вещества сульфаниламиды являются конкурентами р-аминобензойной кислоты (ПАБК). ПАБК используется бактериями для синтеза фолиевой кислоты — фактора их роста. Сульфаниламидные препараты тормозят ферментативную стадию на пути синтеза фолиевой кислоты благодаря конкуренции ПАБК и синтетического аналога. Торможение синтеза фолиевой кислоты в присутствии сульфаниламидов обусловливает бактериостатический эффект последних. Поскольку человек не синтезирует фолиевую кислоту из ПАБК и других метаболитов (см. разд. 10.3), сульфаниламидные препараты в лечебных дозах существенно не влияют на его организм (однако подавляют жизнедеятельность полезной микрофлоры кишечника, которая, в частности, является источником ряда витаминов для человека; поэтому лечение сульфаниламидными препаратами — сульфадимезином» этазолом, сульфамонометоксином и т. д. сопровождается приемом поливитаминов).
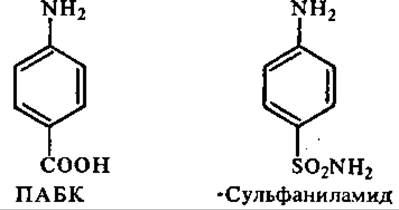
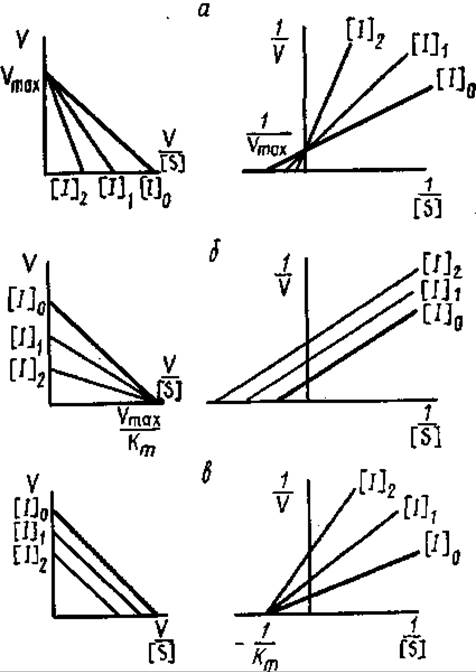
Рис. 3.21. Графики Эди-Хофсти (слева) и Лайнуивера-Бэрка (справа) для различных типов ингибирования: а — конкурентное, б — бесконкурентное, в — неконкурентное