Практическая химия белка - А. Дарбре 1989
Применение масс-спектрометрии электронного удара для определения аминокислотной последовательности пептидов и белков
Общие принципы масс-спектрометрического определения аминокислотной последовательности пептидов и белков
Главное достоинство масс-спектрометрического определения аминокислотной последовательности заключается в возможности анализа смесей пептидов [24, 25], в противном случае масс-спектрометрия не могла бы конкурировать с классическими методами установления первичной структуры пептидов и белков, потому что ее использование обходится намного дороже, чем применение ручного дансил-эдмановского метода анализа, чувствительность которого порой оказывается более высокой. Химикам-белковикам хорошо известно, что процесс выделения индивидуальных пептидов из смеси, образующейся при ферментативном расщеплении белков, является именно той стадией, которая лимитирует скорость определения аминокислотной последовательности пептидов классическими методами. Несмотря на то что первичная структура крупных пептидов в настоящее время обычно устанавливается автоматически с применением секвенаторов, для нахождения мест встраивания этих фрагментов в белковую цепь всегда приходится подвергать молекулу белка более глубокому неспецифическому расщеплению с образованием многих небольших пептидов. Для того чтобы установить аминокислотную последовательность этих коротких пептидов ручным способом, необходимо предварительно разделить смесь на индивидуальные компоненты. При определении структуры этих пептидов методом масс-спектрометрии необходимость выделения отдельных компонентов отпадает вовсе, а в случае сложных смесей достаточно грубого разделения смеси на отдельные фракции.
Исследуемую фракцию после модификации описанным выше способом помещают в испаритель и посредством штока вводят в ионный источник. Благодаря различной летучести модифицированных пептидов при плавном нагревании из смеси дробно возгоняются индивидуальные компоненты, и поэтому масс-спектры, которые записываются в определенные промежутки времени, обычно характеризуют те пептиды, содержание которых в газовой фазе является преимущественным. Сравнивая масс-спектры образца, получаемые при различных температурах, удается однозначно определять аминокислотную последовательность индивидуальных пептидов смеси. Такой подход дает хорошие результаты при анализе смесей, содержащих до пяти различных пептидов. Ниже будут обсуждены общие приемы установления первичной структуры белков, которые позволят наиболее эффективно использовать преимущества масс-спектрометрии при установлении аминокислотной последовательности; коротких пептидов. При этом следует помнить, что для достижения максимального эффекта при установлении первичной структуры неизвестного белка всегда следует комбинировать классические и масс-спектрометрический методы. Такой подход может выглядеть следующим образом:
1) N-концевая аминокислотная последовательность белка или крупных пептидов определяется с помощью автоматических секвенаторов;
2) пептиды, образующиеся в результате специфического ферментативного расщепления белка трипсином или химотрипсином, структурно анализируются ручным дансил-эдмановским методом;
3) аминокислотная последовательность коротких пептидов, образующихся при неспецифическом расщеплении молекулы белка эластазой или субтилизином с целью установления мест встраивания крупных пептидов в белковую цепь, определяется масс-спектрометрически при анализе их смесей [2, 4, 25].
В ряде случаев как метод решения структурных задач следует предпочесть масс-спектрометрию. Например, для определения нуклеотидной последовательности участка ДНК, соответствующего определенному гену, потребуется затратить несравненно меньше усилий, если каким-либо методом определена частичная аминокислотная последовательность белка, первичную структуру которого он кодирует. Такая информация может быть получена в течение нескольких недель с использованием масс-спектрометрии. Масс-спектрометрия — идеальный метод быстрой оценки степени гомологичности родственных белков [4]. Кроме того, масс-спектрометрия — это и непревзойденный метод структурного анализа амидов и определения положения остатков триптофана в пептидной цепи. План эксперимента при определении аминокислотной последовательности с применением масс- спектрометрии можно описать следующим образом:
1) белок неспецифически расщепляется эластазой или субтилизином. Глубина гидролиза проверяется аналитическим электрофорезом при pH 6,5;
2) основная масса коротких пептидов отделяется от негидролизованного белка и крупных пептидов гель-фильтрацией;
3) смесь коротких пептидов разделяется колоночной хроматографией на катионитах, например на дауэксе [4, 25], или высокоэффективной жидкостной хроматографией. Наилучшее разделение на катионитах достигается при элюировании смесью пиридина с уксусной кислотой [4, 25], а при высокоэффективной жидкостной хроматографии — смесью уксусной кислоты с пропанолом [26]. Присутствие пептидов в элюенте устанавливается аналитическим электрофорезом (для анализа используется ~ 1/100 каждой фракции);
4) в результате отбора определенных фракций и их высушивания готовятся пробы, содержащие в смеси до пяти пептидов;
5) пробы химически модифицируются и анализируются методом масс-спектрометрии.
Применение такого подхода позволяет опытному исследователю примерно в течение двух месяцев получить 70% информации о аминокислотной последовательности белка с М≤20 000.
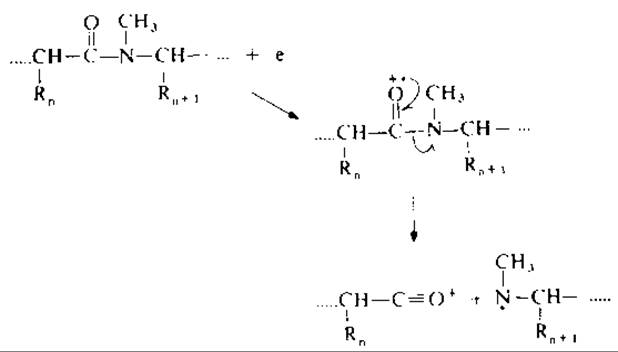
РИС. 19.3. С—N-Разрыв пептидных связей в перметилированных пептидах.