Основы биохимии - Филиппович Ю. Б. 1999
Липиды и их обмен
Обмен липидов
Обмен жиров (триглицеридов). Гидролиз жиров. Первой фазой обмена жиров (триглицеридов) является гидролиз, в результате которого освобождаются глицерин и высшие жирные кислоты. Реакция гидролиза триглицеридов ускоряется гидролазой эфиров глицерина — липазой.
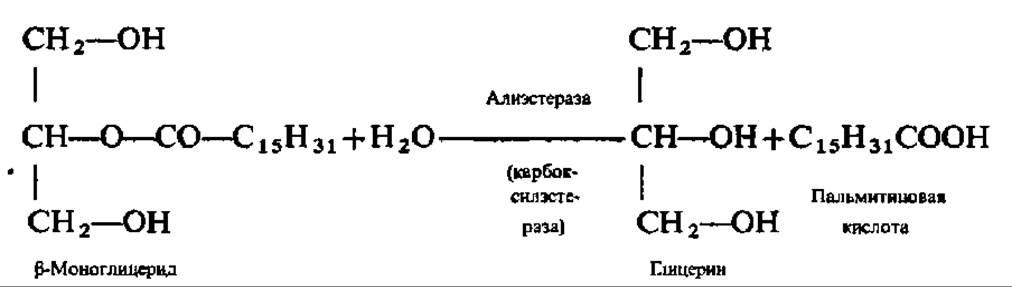
Гидролиз триглицеридов идет ступенчато: сначала распадаются две внешние сложноэфирные связи (a-связи). Уравнение этой реакции приведено выше (см. с. 129). Так осуществляется, например, гидролиз триглицеридов в кишечнике человека и животных при каталитическом воздействии липазы поджелудочной железы (М = 48000, мономер). ß-Моноглицериды всасываются стенкой кишечника и либо идут на ресинтез триглицеридов уже в кишечной стенке, либо распадаются далее под действием неспецифических эстераз, способных ускорять реакции гидролиза сложных эфиров вторичных спиртов. Примером может служить гидролиз ß-моноглицерида в присутствии алиэстеразы печени:
В растительном царстве липазы широко распространены в семенах и вегетативных органах растений. Специфичность их к а- и ß-глицеридам не установлена, а из дрожжевых грибков выделена липаза, атакующая в равной мере и а- и ß-связи (М = 55000, мономер, содержит 7% углеводов). Среди липаз из микроскопических грибов зафиксированы множественные формы. Различают простые липазы, которые каталитически ускоряют освобождение высших жирных кислот из свободных триглицеридов, и липопротеинлипазы, способствующие гидролизу связанных с белками липидов.
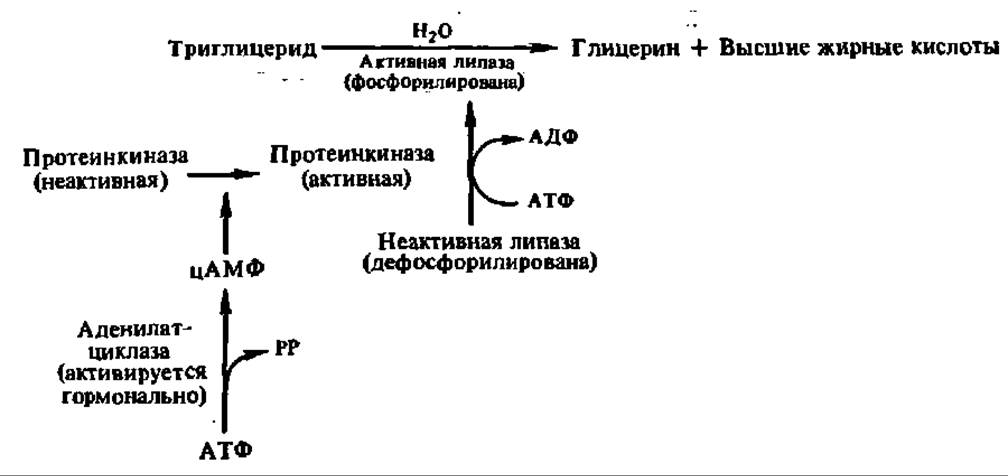
Третичная структура дрожжевой липазы выяснена. Ее полипептидная цепь (430 аминокислотных остатков) сложена в глобулу (7x7x5 нм), в центре которой находится активный центр, включающий остаток гистидина. Высказаны предположения и о структуре активного центра панкреатической липазы: ведущую роль в нем играют радикалы гистидина, серина, дикарбоновых аминокислот и изолейцина. Как и в случае других гидролаз, радикал гистидина служит для переноса протонов, а радикал серина — для акцептирования ацильной группы, высвобождающейся в момент распада сложноэфирной связи в молекуле триглицерида. Радикал изолейцина взаимодействует с углеводородным радикалом остатка высшей жирной кислоты и способствует закреплению молекулы триглицерида в активном центре фермента (рис. 122). Выяснено, что активность липаз регулируется путем их фосфорилирования — дефосфорилирования:
Распад глицерина и высших жирных кислот. В обмене жиров характерно широкое использование продуктов их распада для ресинтеза. Поэтому значительная часть ß-моноглицеридов, глицерина и свободных высших жирных кислот, освобождающихся при гидролизе триглицеридов, используется для ресинтеза триглицеридов же, но несколько иного состава и строения, характерного для того или иного организма (если для этого используются пищевые жиры) или органа (если идет перестройка жиров в пределах организма).
Так как новообразованные жиры неизбежно отличаются от распавшихся триглицеридов по строению и соотношению остатков высших жирных кислот (в соответствии с их видовой или тканевой специфичностью), то часть высших жирных кислот и некоторая доля глицерина подвергаются дальнейшей деструкции.
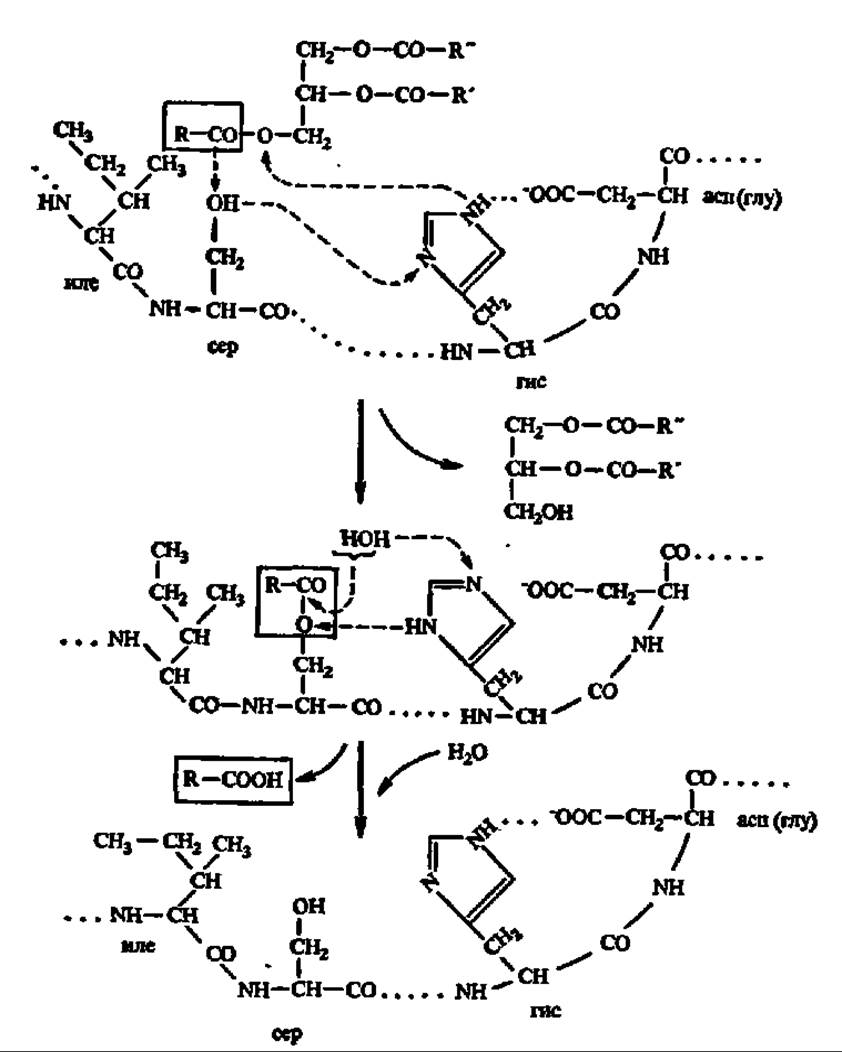
Рис. 122. Механизм гидролиза триглицеридов (пояснения в тексте)
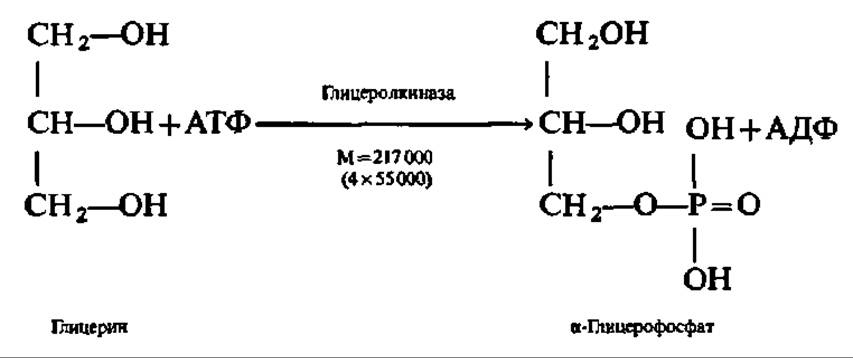
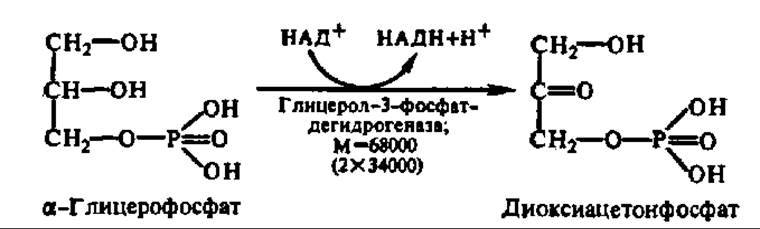
Глицерин независимо от того, поступил ли он на ресинтез жиров или будет претерпевать дальнейший распад, прежде всего фосфорилируется. Донором остатка фосфорной кислоты в этой реакции служит АТФ. Процесс ускоряется соответствующей фосфотрансферазой:
Глицерофосфат в основном идет на синтез новых молекул триглицеридов, но часть его окисляется с образованием диоксиацетон-фосфата:
Диоксиацетонфосфат изомеризуется в 3-фосфоглицериновый альдегид, который затем вступает в обменные реакции, рассмотренные ранее (см. гл. VIII).
Наибольший интерес в процессах обмена продуктов гидролиза триглицеридов представляет судьба высших жирных кислот. Первые гипотезы относительно механизма их распада были высказаны в начале нашего столетия (Ф. Кнооп, 1904). В дальнейшем они были уточнены и развиты благодаря работам лабораторий Ф. Линена, Д. Ірина, С. Очоа, Г. Ларди и А. Ленинджера. Современные данные по этому вопросу сводятся к следующему. Считают, что высшие жирные кислоты разрушаются преимущественно путем ß-окисления. Ненасыщенные высшие жирные кислоты (олеиновая, линолевая, линоленовая и др.) предварительно восстанавливаются до предельных кислот. Окисление предельных высших жирных кислот осуществляется ступенчато, путем отщепления от их молекул двууглеродных фрагментов. Все реакции многостадийного окисления ускоряются специфическими ферментами, причем начиная с третьей фазы (см. ниже) они собраны в виде метаболона с М = 260 000 Да.
Первая фаза распада высших жирных кислот заключается в активировании их путем образования соединения с коэнзимом А (КоА), содержащего макроэргическую связь. Последняя, видимо, способствует более гладкому протеканию реакций окисления образовавшегося соединения, которое называют ацил-коэнзимом А (ацил-КоА). Взаимодействие высших жирных кислот с КоА ускоряется специфическими лигазами — ацил-КоА-синтетазами трех видов специфичных соответственно для кислот с коротким, средним и длинным углеводородными радикалами. Они локализованы в мембранах эндоплазматической сети и в наружной мембране митохондрий. По-видимому, все ацил-КоА-синтетазы являются мультимерами; так, фермент из микросом печени имеет М = 168 000 и состоит из 6 идентичных субъединиц с М = 28000.
Уравнение реакции активирования высших жирных кислот перед их окислением таково:
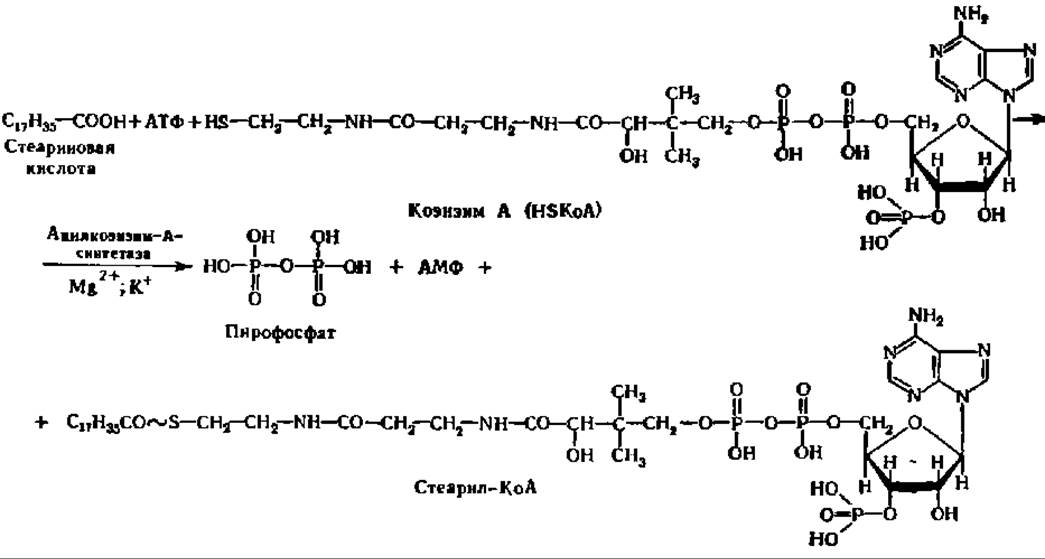
Образующийся в этой реакции пирофосфат энергично расщепляется до Н3РО4 при участии пирофосфатазы, что обеспечивает смещение равновесия всего процесса вправо.
Вторая фаза распада высших жирных кислот состоит в окислении ацил-КоА при посредстве ацил-КоА-дегидрогеназы, содержащей флавинадениндинуклеотид (ФАД, см. с. 120) в качестве кофермента:

Существует, по меньшей мере, три ацил-КоА-дегидрогеназы, предпочитающие короткие, средние и длинные ацильные радикалы соответственно.
Третья фаза окисления высших жирных кислот состоит в присоединении молекулы воды по месту двойной связи дегидроацил-КоА. Эта реакция ускоряется соответствующими гидролиазами. Так как присоединение воды (гидратация) идет по двойной связи (двойная связь условно обозначается частицей ен), то эти ферменты по современной номенклатуре называют еноил-КоА-гидратазами. Один из них специфичен к цисформам дегидроацил-КоА, другой — к транс-формам:
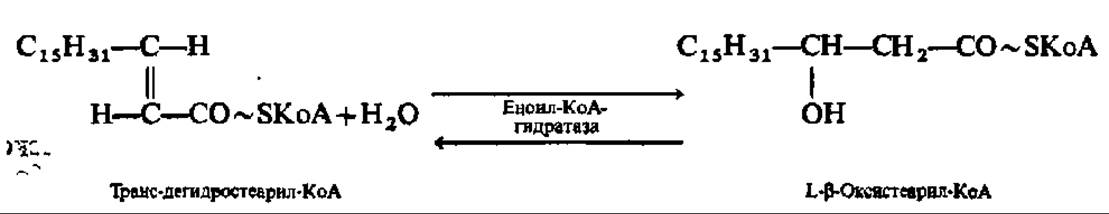
Четвертая фаза распада высших жирных кислот заключается в еще одном окислении путем отнятия двух атомов водорода от ß-углеродного атома (отсюда и весь рассматриваемый здесь механизм носит название ß-окисления). Как и во второй фазе процесса, снятие атомов водорода осуществляется оксидоредуктазой, но с НАД+ в качестве кофермента. Фермент специфичен лишь к L-форме ß-оксиацил-КоА:
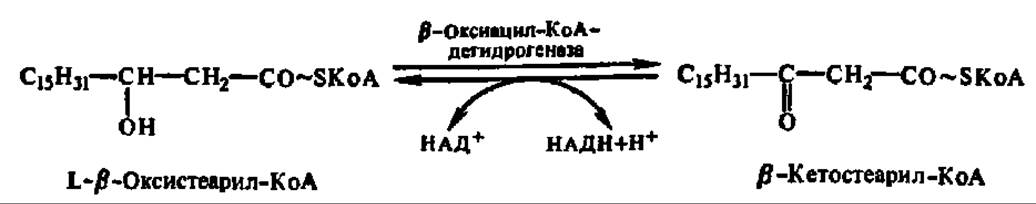
Наконец, последняя, пятая фаза распада сводится к переносу предобразованной в молекуле ß-кетоацил-КоА новой ацильной группировки на молекулу КоА. Этот процесс ускоряется соответствующей ацилтрансферазой, которую предпочитают называть тиолазой, так как сама реакция, по существу, представляет расщепление С—С-связи с присоединением по месту разрыва элементов HS-группы (тиолиз):
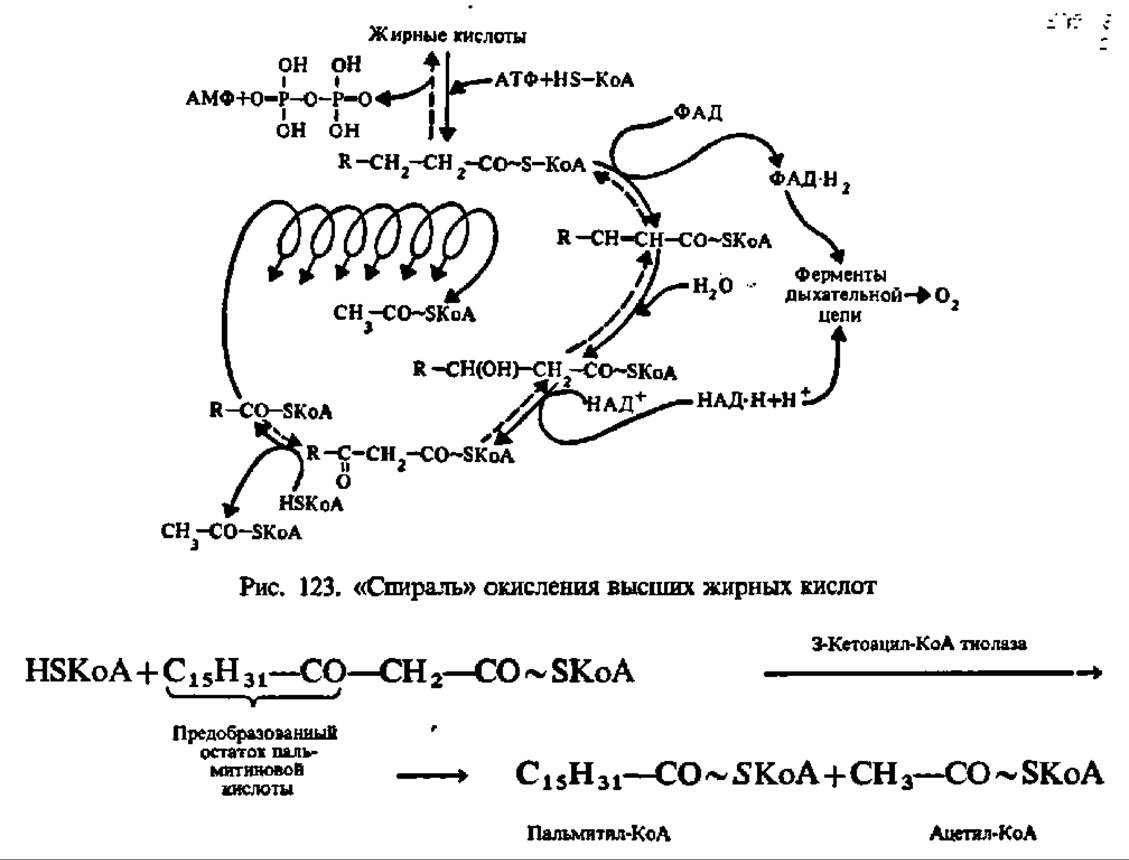
В результате описанных выше реакций молекула высшей жирной кислоты (стеариновой в нашем примере) укорачивается на два углеродных атома и образуются пальмитиновая и уксусная кислоты в виде производных КоА (пальмитил- и ацетил-КоА). Этот процесс многократно повторяется (рис. 123). Окончательным продуктом ß-окисления высших жирных кислот с четным числом углеродных атомов является ацетил-КоА, а с нечетным — пропионил-КоА.
Если бы ацетил-КоА накапливался в организме, то запасы HSKoA скоро исчерпались бы и окисление высших жирных кислот остановилось. Но этого не происходит, так как КоА быстро освобождается из состава ацетил-КоА. К этому приводит ряд процессов: ацетил-КоА включается в цикл трикарбоновых и дикарбоновых кислот (см. рис. 117) или весьма близкий к нему глиоксилевый цикл (см. ниже), или, наконец, ацетил-КоА используется для синтеза полициклических спиртов (стеролов) и соединений, содержащих изопреноидные группировки и т. п.
ß-Окисление высших жирных кислот протекает в митохондриях. Естественно, что, поскольку в них же локализованы ферменты дыхательного цикла, ведущие передачу атомов водорода и электронов на кислород сопряженно с окислительным фосфорилированием, ß-окисление высших жирных кислот может явиться источником энергии для синтеза АТФ.
В некоторых случаях высшие жирные кислоты представляют единственные вещества, окисление которых служит источником энергии для окислительного фосфорилирования (биосинтез белка в шелкоотделительной железе шелкопряда, полет насекомых).
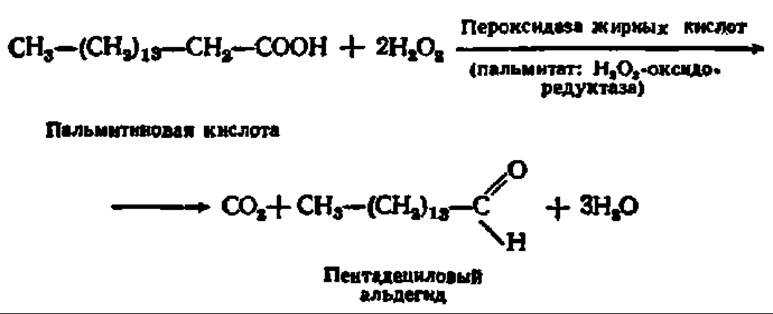
В бесструктурной части клеточного содержимого тоже есть ферментные системы, способные окислять высшие жирные кислоты. Окисление, идущее здесь, осуществляется по а-углеродному атому и называется а-окислением. В нем принимают участие Н2О2 и фермент — пероксидаза:
Альдегид высшей жирной кислоты окисляется при посредстве дегидрогеназы в высшую жирную кислоту, и процесс повторяется снова:
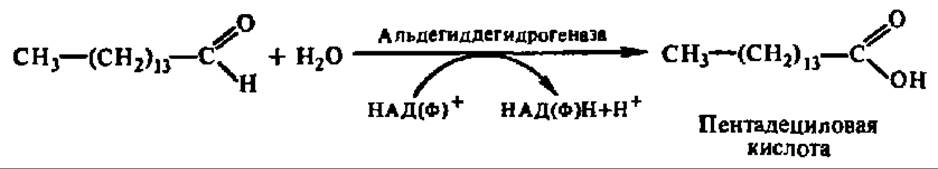
Так укорачиваются цепи высших жирных кислот, содержащих в своем составе от 15 до 18 углеродных атомов. Указанный дополнительный путь а-окисления высших жирных кислот характерен только для растений. Существует также ферментная система, обеспечивающая ω-окисление, т. е. окисление по СН3-группе радикала высших жирных кислот. Она изучена в микросомной фракции печени и у микроорганизмов. Сначала под действием монооксигеназы (см. гл. X) возникает ω-оксикислота, а затем — дикарбоновая высшая жирная кислота. Последняя укорачивается с любого конца посредством реакций ß-окисления.
Обмен ацетил-КоА. Выше отмечалось, что ацетил-КоА быстро расходуется, высвобождая свободный HSKoA. Следовательно, в реакциях ß-окисления высших жирных кислот HSKoA и его ацильные производные, будучи коферментами, выполняют каталитическую функцию.
Одним из процессов, в результате которого осуществляется регенерация HSKoA, является образование ацетоуксусной кислоты. Этот путь преобразования ацетил-КоА широко представлен в митохондриях печени. Сначала две молекулы ацетил-КоА конденсируются с образованием β-кетобутирил-КоА и выделением одной молекулы свободного HSKoA:
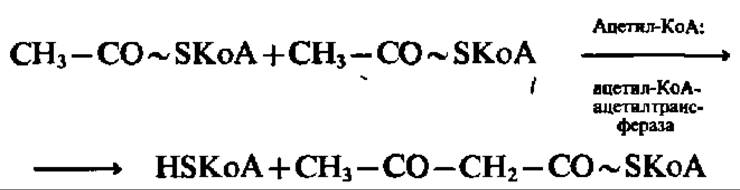
Далее HSKoA высвобождается из состава p-кетобутирил-КоА. Известно несколько реакций, приводящих к такому результату. Среди них преобладает реакция, в которую вовлекается еще одна молекула ацетил-КоА:
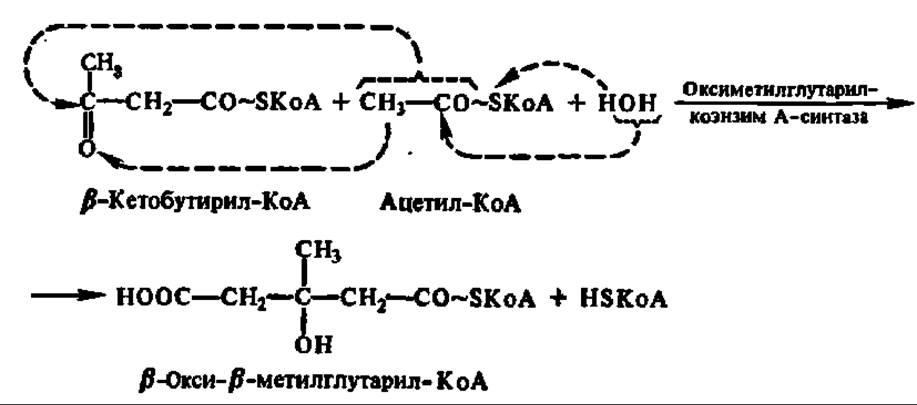
Образовавшийся в результате реакции конденсации ß-окси- ß-метилглутарил-КоА представляет весьма важное соединение, так как из него может синтезироваться мевалоновая кислота — ключевое соединение в синтезе стеролов и изопреноидов (см. с. 402). Однако в данном случае (т. е. в митохондриях печени) ß-окси-β-метилглутарил-КоА распадается на ацетоуксусную кислоту и ацетил-КоА:
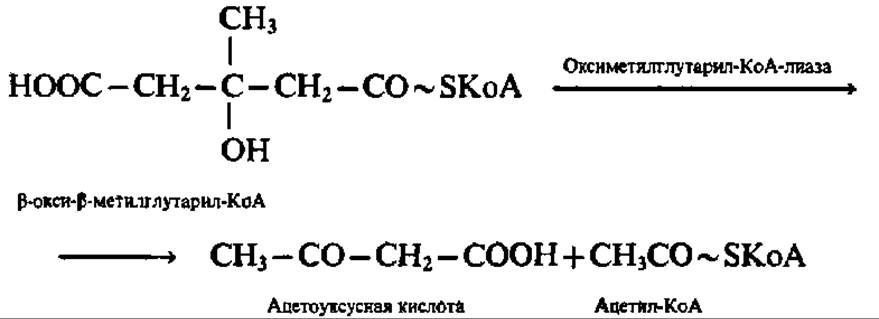
В результате перечисленных выше реакций из двух молекул ацетил-КоА синтезируется одна молекула ацетоуксусной кислоты и высвобождаются две молекулы HSKoA.
Другой распространенный путь обмена ацетил-КоА — это взаимодействие с енольной формой щавелевоуксусной кислоты с образованием цитрил-КоА, т. е. вступление в цикл трикарбоновых и дикарбоновых кислот. При гидролизе цитрил-КоА высвобождается HSKoA, а лимонная кислота обменивается далее в соответствии с ранее рассмотренной схемой (см. рис. 117). Такой путь обмена ацетил-КоА характерен для митохондрий подавляющего большинства тканей — почек, мышц и т. д., за исключением печени.
Известно еще много процессов, которые ведут к высвобождению HSKoA из состава ацетил-КоА. Ацетил-КоА является универсальным донором ацетильных групп для реакций ацетилирования. Существует более десяти специфических ацетилтрансфераз, обеспечивающих ускорение реакций переноса ацетильных остатков (синтез ацетилхолина, N-ацетилглюкозамина и т. п.) Во всех случаях выделяется свободный HSKoA.
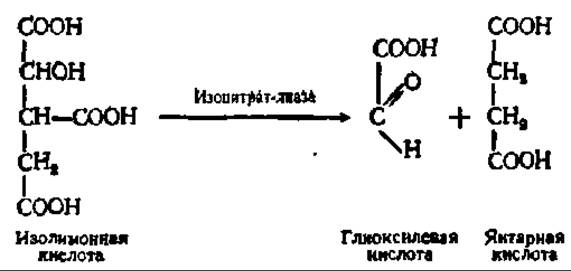
Высвобождение HSKoA из состава ацетил-КоА может сопровождаться накоплением щавелевоуксусной кислоты. Это происходит в том случае, когда ацетил-КоА обменивается при посредстве так называемого глиоксилевого цикла. В значительной мере химические процессы, протекающие при осуществлении глиоксилевого цикла, совпадают с таковыми цикла дикарбоновых и трикарбоновых кислот (см. рис. 117). Все идет одинаково, вплоть до образования изолимонной кислоты. Однако в глиоксилевом цикле изолимонная кислота расщепляется на янтарную и глиоксилевую кислоты:
Янтарная кислота тем же путем, как и в цикле дикарбоновых и трикарбоновых кислот, превращается в щавелевоуксусную кислоту. Глиоксилевая же кислота конденсируется с новой молекулой ацетил-КоА и образует в конечном счете свободный HSKoA и яблочную кислоту:
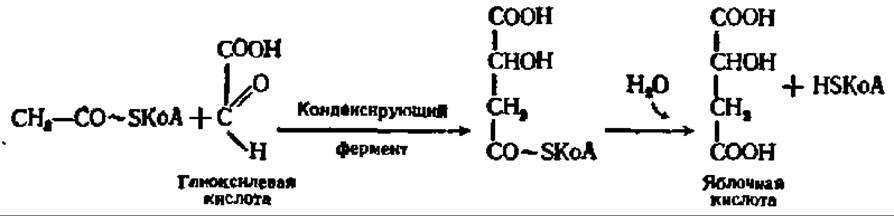
Последняя, дегидрируясь, дает начало одной молекуле щавелевоуксусной кислоты. Таким образом, при помощи глиоксилевого цикла ацетил-КоА превращается в щавелевоуксусную кислоту и свободный HSKoA. Этот процесс имеет огромное значение для обеспечения в организме синтеза углеводов за счет распадающихся высших жирных кислот.
Обмен пропионил-КоА. Пропионил-КоА, являющийся конечным продуктом ß-окисления высших, жирных кислот с нечетным числом углеродных атомов, превращается в сукцинил-КоА путем двух последовательных реакций:
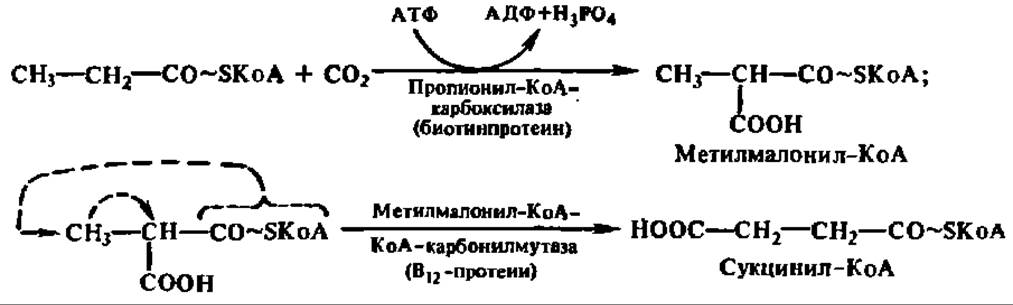
Далее сукцинил-КоА утилизируется через цикл трикарбоновых и дикарбоновых кислот.
Синтез высших жирных кислот. Долгое время считали, что синтез высших жирных кислот идет путем обращения реакции ß-окисления высших жирных кислот. Однако после того как было обнаружено, что для его осуществления необходим не только ацетил-КоА, но и СО2 (из которых при посредстве АТФ-зависимой реакции возникает малонил-КоА), а сам процесс ускоряется синтетазой высших жирных кислот, локализованной в растворимой фракции клетки, эта точка зрения была оставлена. В 60-е годы огромную роль в расшифровке механизма биосинтеза высших жирных кислот сыграли работы Ф. Линена и сотр.
Современные представления о биосинтезе высших жирных кислот в организме представлены на схеме 10.
Начальный этап биосинтеза высшей жирной кислоты, приводящий к синтезу малонил-КоА, ускоряется полифункциональным ферментом (М = 225000 Да), содержащим домен биотинкарбоксилазы, биотин-карбоксил-проводящий домен и домен транскарбоксилазы. Первый домен обеспечивает ускорение реакции карбоксилирования биотина (рис. 124), который через радикал лизина присоединен ко второму, биотин-карбоксилпроводящему домену. Обладая высокой степенью подвижности, карбоксилированный биотин переносит СО2 в активный центр третьего домена — транскарбоксилазы, который снимает с него СО2 и непосредственно передает его на ацетил-КоА, образуя малонил-КоА:
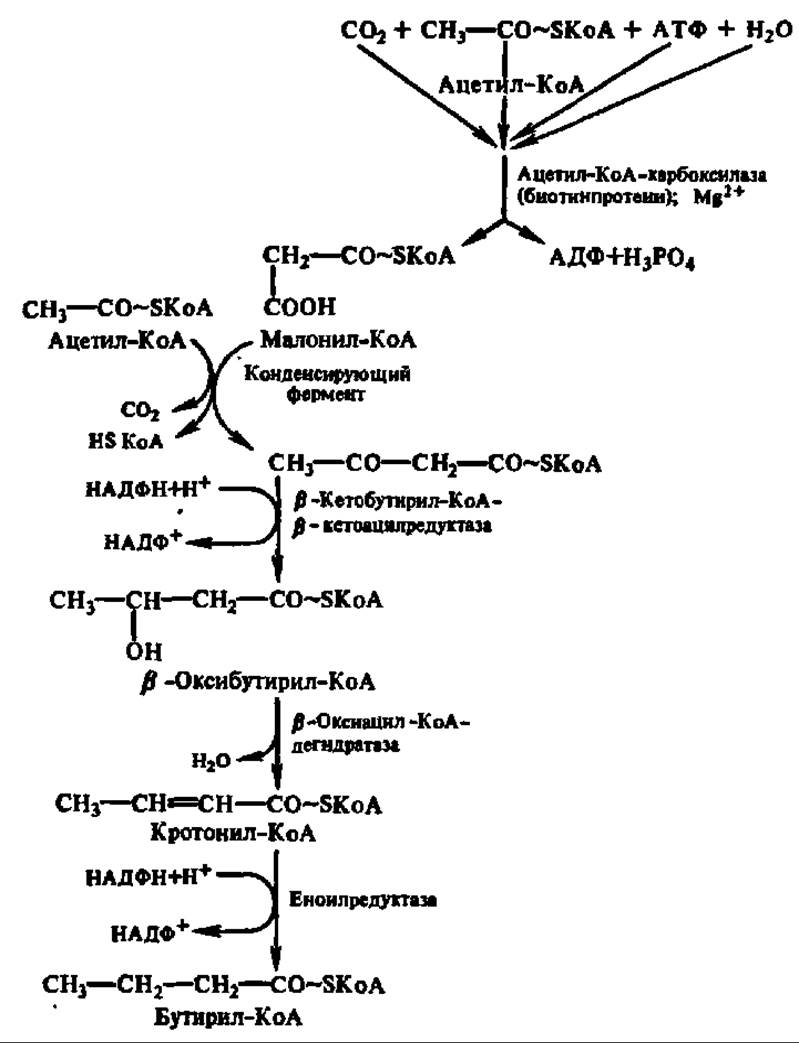
Схема 10. Механизм биосинтеза высших жирных кислот Многократное повторение наращивания радикала на два атома углерода приводит к синтезу кислот, содержащих 16 и более углеродных атомов.
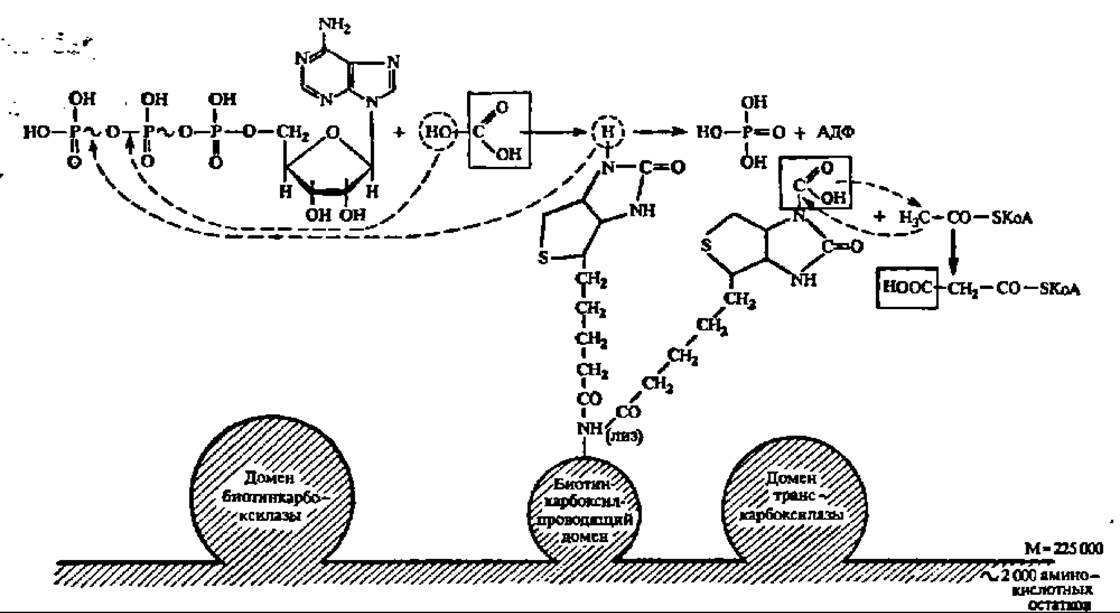
Рис. 124. Механизм биосинтеза малонил-КоА (пояснения в тексте)
В мономерном состоянии ацетил-КоА-карбоксилаза не активна и приобретает способность карбоксилировать CH3CO~SKoA только после соединения мономеров в нитевидный олигомер с молекулярной массой в несколько сотен миллионов и длиной около 500 нм. Процесс олигомеризации регулируется аллостерически присоединением лимонной кислоты.
Кроме того, активность ацетил-КоА-карбоксилазы регулируется ее фосфорилированием (снижение) и дефосфорилированием (повышение). Таким образом, интенсивность работы цикла трикарбоновых и дикарбоновых кислот и уровень протеинкиназных и протеинфосфатазных реакций предопределяет объем биосинтеза высших жирных кислот, последующие стадии которого осуществляются при посредстве второго полифункционального энзима — синтетазы высших жирных кислот.
Этот комплекс у высокоорганизованных форм (млекопитающие, птицы, насекомые) характеризуется М = 400 000—560000, а у низкоорганизованных (микобактерии, низщие грибы, жгутиковые) — 1,4 ∙ 06 — 2,3 ∙ 106. В нем сосредоточены все каталитические активности, необходимые для обеспечения многоступенчатого биосинтеза высших жирных кислот, а также ацилпроводящий домен, функция которого состоит в передвижении ацильной группы от одного субдомена к другому в строгом соответствии с химизмом этого процесса. Представление о работе синтетазы высших жирных кислот дают рис. 125 и 126.
В первом случае (рис. 125, синтетаза из печени цыпленка) каждая его полипептидная цепь длиной около 2300 аминокислотных остатков образует 3 домена и 8 субдоменов, с каждым из которых связана определенная функция. Однако один из субдоменов, а именно — обладающий ß-кетоацил-синтетазной активностью, работает только в паре с другой такой же полипептидной цепью, расположенной по отношению к первой по правилу «голова к хвосту». Он перебрасывает ацетильную (первый цикл синтеза) или ацильную (последующие циклы) группу со своего остатка цистеина (рис. 125) на малонильный остаток, закрепленный на HS-группе пантотеиновой «руки» ацил-переносящего субдомена соседней субъединицы. Возникший ß-кетоацильный остаток при помощи той же пантотеиновой руки перемещается по остальным трем субдоменам домена II (восстанавливающего ß-кетоацил в ацил). Далее ацетил (ацил) трансферазный домен элонгационного домена I посылает эту ацильную группу на HS-группу остатка цистеина 3-кетоацил-синтетазного субдомена и начинается новый цикл удлинения цепи, но уже на соседней субъединице синтетазы высших жирных кислот. По достижении ацильным радикалом длины в 16 атомов углерода он отщепляется тиоэстеразой в виде ацил-КоА.
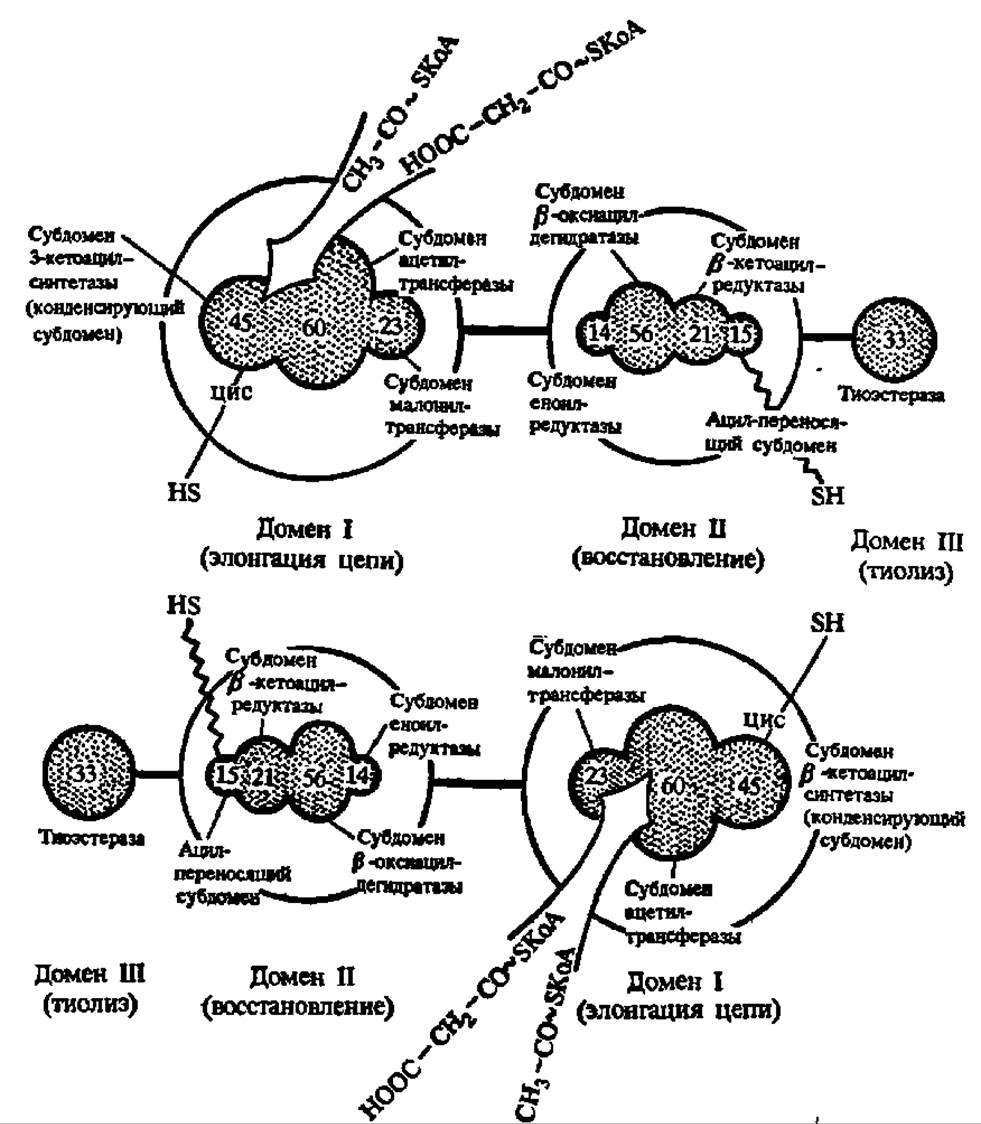
Рис. 125. Строение и механизм действия синтетазы высших жирных кислот печени цыпленка:
цифрами обозначены молекулярные массы (в кДа) субдоменов. Остальные пояснения — в тексте
Во втором случае (синтетаза из дрожжей) принцип согласованной и взаимозависимой работы субъединиц синтетазы высших жирных кислот остается в силе (рис. 126, пояснения в подрисуночной подписи).
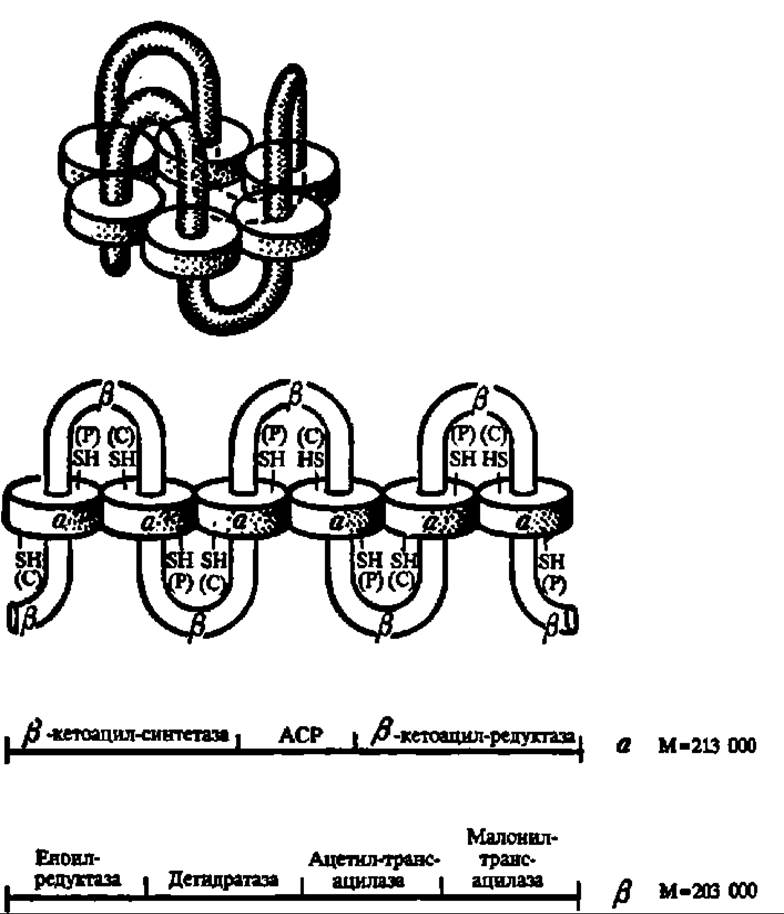
Рис. 126. Структура синтетазы высших жирных кислот дрожжей
Фермент представляет а6 ß6 комплекс с М = 2469000 Да. Каждая из субъединиц обладает своим набором каталитических активностей. АСР — ацилпереносящий домен, обладающий пантотеиновой «рукой» [обозначена SH (Р)]. SH— (С) — цистеиновый остаток ß-кето-ацилсинтетазы, с которого идет перенос ацетильной (первый цикл) и ацильных (последующие циклы) групп па малонильный остаток, закрепленный на HS-гpyппe пантотеиновой «руки». Вверху — свернутая; внизу — развернутая форма фермента. Механизм действия полностью аналогичен таковому синтетазы высших жирных кислот из печени цыпленка
Синтез триглицеридов. Из глицерина и высших жирных кислот при каталитическом воздействии липазы in vitro удается получить триглицерид. В связи с этим полагали, что и in vivo липаза может проявлять не только гидролитическое, но и синтезирующее действие и что с ее помощью могут возникать триглицериды путем обращения реакции гидролиза. В последние годы доказана принципиально иная схема биосинтеза триглицеридов, в которой отправными веществами служат ацил-КоА и фосфоглицерин, а ферментами — ацилтрансферазы. Учитывая общую тенденцию в эволюции наших представлений в сторону совершенно четкого разграничения и определенного различия путей распада и синтеза основных классов органических соединений в биологических объектах (см. синтез и распад белков, нуклеиновых кислот, полисахаридов), несомненно, что синтез жиров путем обращения гидролиза вряд ли широко представлен в природе и что главный путь новообразования триглицеридов заключается в осуществлении реакций трансацилирования.
Как упомянуто выше, исходными веществами при синтезе триглицеридов посредством реакций трансацилирования являются а-фосфоглицерин и разнообразные ацил-КоА. Первый возникает при фосфорилировании глицерина или при восстановлении фосфодиоксиацетона. Прямое фосфорилирование глицерина характерно для почек животных, а также микроорганизмов. Восстановление фосфодиоксиацетона идет в мышцах, слизистой кишечника и т. п. Вторые синтезируются либо путем активирования высших жирных кислот, либо путем новообразования из ацетил-КоА (см. выше). Вначале посредством реакций трансацилирования синтезируется фосфатидная кислота:
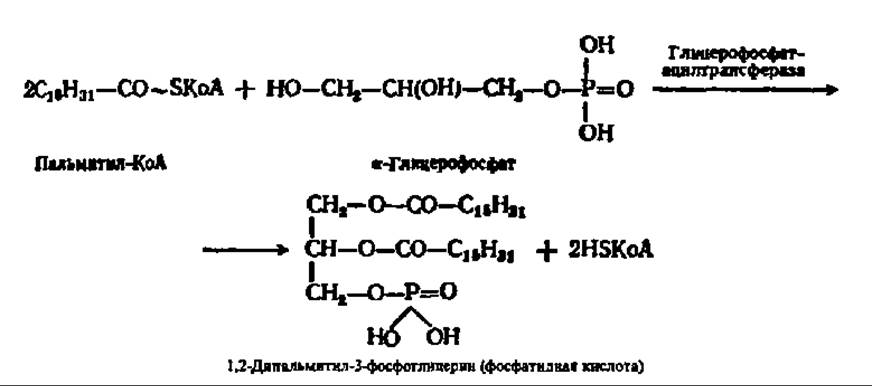
При участии фосфатазы фосфатидная кислота гидролизуется с образованием диглицерида и фосфорной кислоты:

Диглицерид вступает снова во взаимодействие с ацил-КоА и образуется триглицерид. И эта реакция ускоряется трансацилазой:
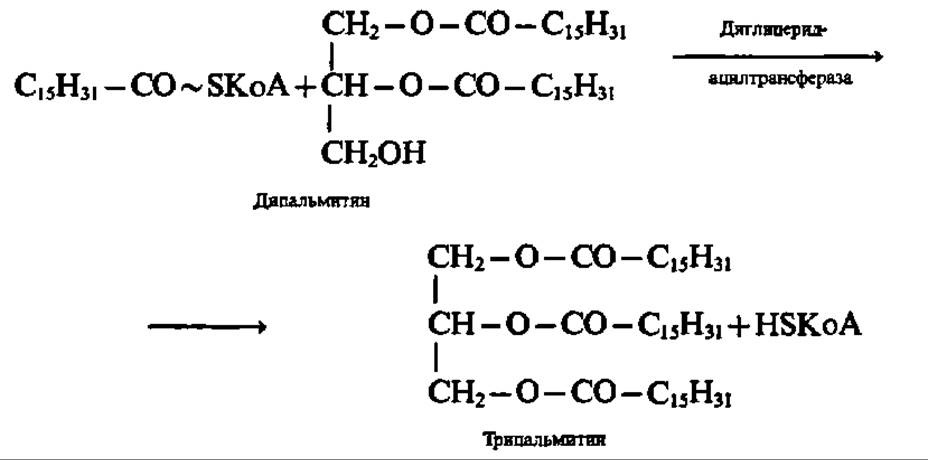
Ферменты, ускоряющие синтез триглицеридов в соответствии с приведенными выше уравнениями, найдены в печени, слизистой оболочке кишечника, жировой ткани и т. п. Интересной особенностью всех указанных ферментов является их липопротеиновая (за исключением глицеролкиназы) природа. Они ведут синтез триглицеридов на мембранах эндоплазматической сети клетки. По мере своего возникновения триглицериды мигрируют и поглощаются жировыми включениями клетки. Из тканей, активно синтезирующих триглицериды (например, печень), они переходят в ткани, где нет активного синтеза (например, кровь). В организме животных существует обычно несколько жировых депо с медленно обменивающимися триглицеридами.
Механизм биосинтеза триглицеридов через фосфатидные кислоты в качестве метаболитов не является единственным. В слизистой оболочке кишечника синтез триглицеридов идет из ß-моноглицеридов при посредстве весьма активной моноглнцеридтрансацилазы:

Само собой разумеется, что диглицерид превращается далее в триглицерид при каталитическом участии диглицеридтрансацилазы (см. предыдущее уравнение). Моноглицеридный тип биосинтеза энергетически вдвое выгоднее фосфатидного пути. Кроме того, недавно обнаружена диоксиацетонфосфатацил-трансфераза, которая может открывать еще один путь биосинтеза ацилглицеринов.
Обмен стеридов. Вступая на путь распада, стериды сразу же гидролизуются на жирную кислоту и стерол. Поскольку стериды представляют собой в химическом отношении сложные эфиры высших жирных кислот и полициклических спиртов (стеролов), то реакция гидролиза ускоряется холестеролэстеразой, действующей также на сложные эфиры других стеролов (см. с, 381).
Холестеролэстераза выделена из поджелудочной железы ряда животных и человека; она представлена мономером с М = 65000—69000, склонным к олигомеризации (М = 300 000—800 000).
Что касается высших жирных кислот, высвобождающихся из состава стеридов при гидролизе, то они далее могут либо использоваться для ресинтеза липидов, в том числе и стеридов, либо распадаться до ацетил-КоА и потом — до СО2 и Н2О. Поэтому рассмотрим, как протекает далее обмен стеролов — второго компонента, образующегося при гидролизе стеридов.
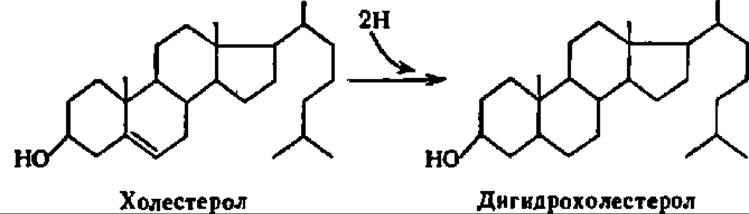
Распад стеролов. Та часть стеролов, которая не используется для ресинтеза стеридов, подвергается видоизменению. Простейшее видоизменение состоит в восстановлении стеролов по двойным связям. Так, холестерол у человека и высших животных превращается в дигидрохолестерол (холестанол), который в виде конформера (копростанола — см. с. 380) выводится из организма:
Более сложный характер носит видоизменение стеролов путем окисления. Сначала появляются ОН-группы в положениях 7 и 12 циклопентанопергидрофенантренового цикла, а затем окисляется боковая цепь, в которой возникает СООН-группа (положение 24). В результате образуются холевые кислоты. Подсчитано, что до 80% холестерола превращается в печени в различные холевые кислоты. При более полном окислении стеролов могут возникнуть стероидные гормоны (см. с. 444). Таким образом, часть стеролов превращается в процессе окисления в различные соединения, выполняющие в организме важные функции.
Синтез стеролов и стеридов. Механизм биосинтеза стеролов долгое время оставался загадочным, хотя давно было известно, что стеролы беспрепятственно синтезируются у большинства органических форм (исключение составляют, например, насекомые). Лишь применение метода меченых атомов позволило расшифровать этот оказавшийся довольно сложным процесс, основные этапы которого, видимо, совпадают у самых различных организмов.
Синтез стеролов осуществляется из ацетил-КоА в качестве исходного вещества. Первые стадии биосинтеза совпадают с реакциями, которые были описаны выше при рассмотрении обмена ацетил-КоА. Напомним, что в результате двух последовательно протекающих при этом реакции из трех молекул ацетил-КоА образуется одна молекула ß-окси-ß-метилглутарил-КоА. Это соединение ферментативным путем восстанавливается в мевалоновую кислоту; восстановление идет по макроэргической связи и сопровождается выделением свободного HSKoA:
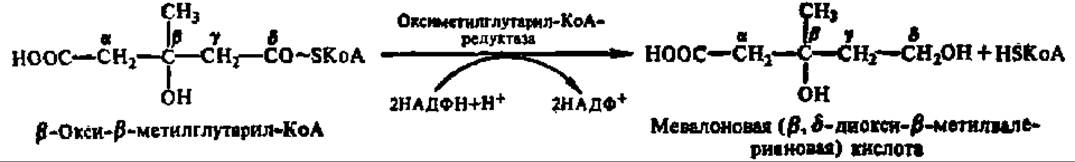
Оксиметилглутарил-КоА-редуктаза из микросом печени крысы имеет М = 32000, но молекулярная масса втрое выше (97092 Да, 887 аминокислотных остатков) у фермента из яичников китайского хомячка. Обе активны только в дефосфорилированном состоянии, тогда как протеинкиназная реакция их полностью инактивирует. Этот фермент считают ключевым при биосинтезе стеролов, так как он успешно конкурирует за субстрат с ферментами других метаболических путей.
Мевалоновая кислота дважды фосфорилируется по 8-гидроксильной группе. Донором остатков фосфорной кислоты в этих реакциях служит АТФ. Процесс ускоряется специфическими фосфотрансферазами:
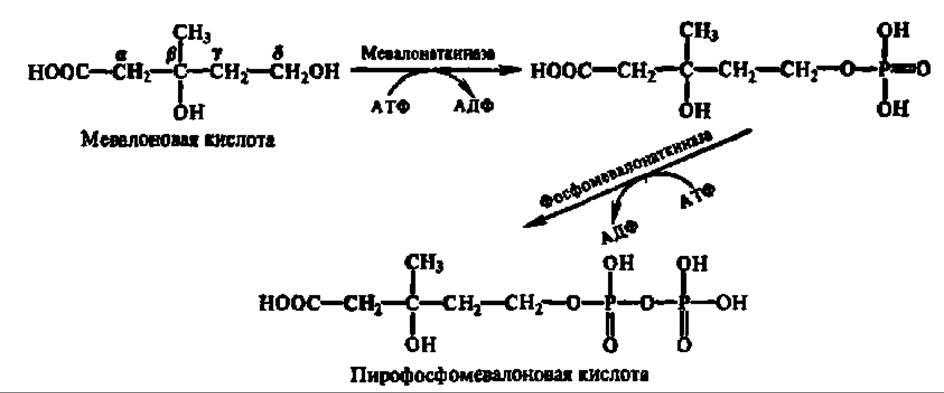
Пирофосфомевалоновая кислота декарбоксилируется. Одновременно осуществляется реакция дегидратирования и образуется изопентенилпирофосфат:
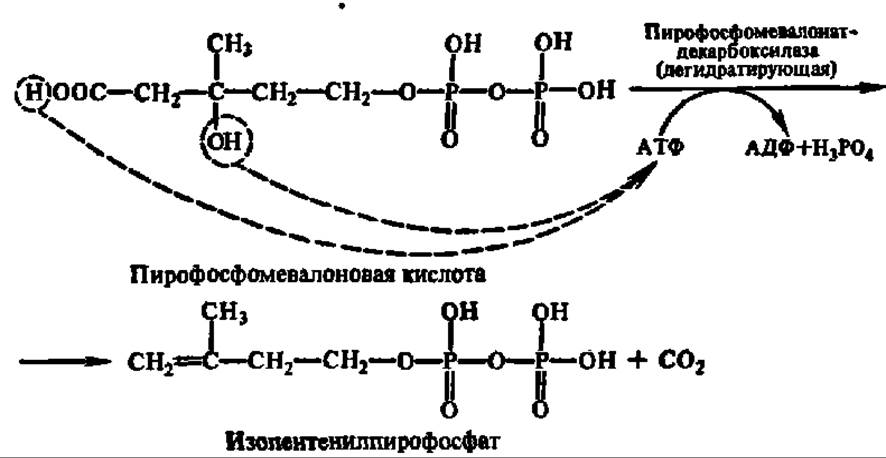
Изопентенилпирофосфат при участии фермента — изопентенилпирофосфат-изомеразы превращается в диметилаллилпирофосфат (см. с. 135).
Из двух вышеупомянутых соединений: изопентенилпирофосфата и диметил-аллилпирофосфата — и идет синтез стеролов. Сначала указанные соединения образуют геранилпирофосфат:
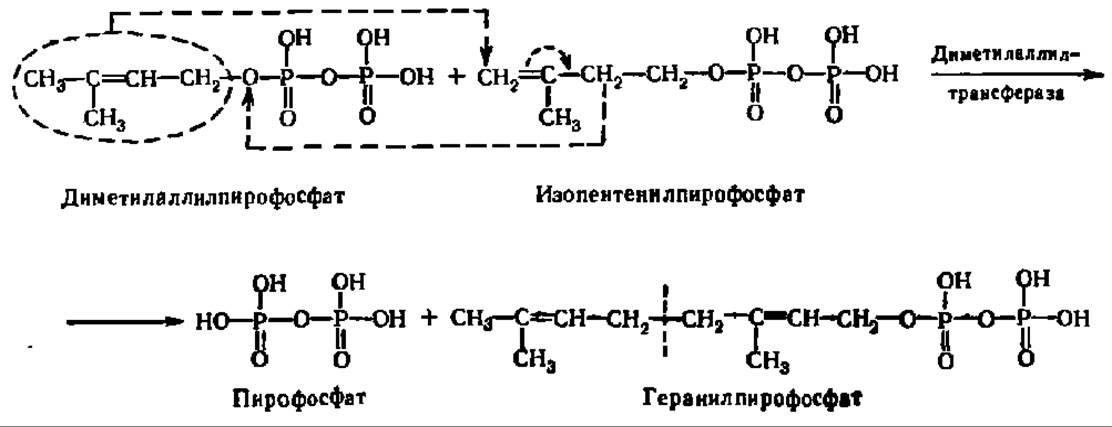
Эта реакция ускоряется ферментом — диметилаллилтрансферазой, обеспечивающей перенос диметилаллильного радикала на раскрывающуюся двойную связь молекулы изопентенилпирофосфата. Одновременно выделяется пирофосфат, получающий атом водорода от близлежащей метиленовой группы. Этот же фермент катализирует реакцию переноса радикала геранила от геранил-пирофосфата к следующей молекуле изопентенилпирофосфата:
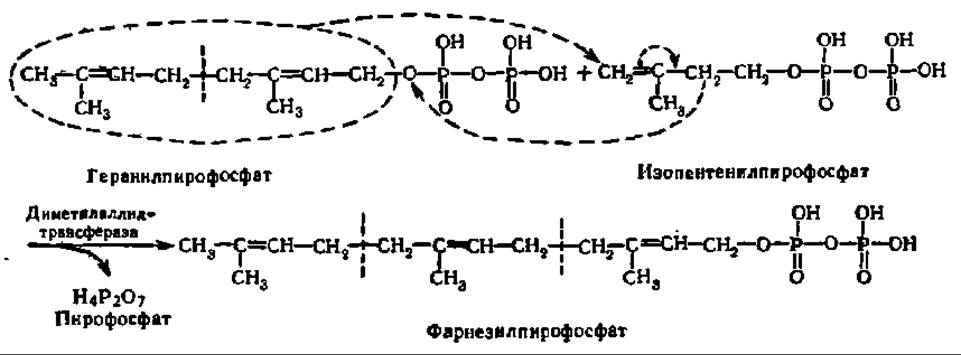
Указанные реакции трансалкилирования поддерживаются сопутствующим гидролизом пирофосфата при участии пирофосфатазы:
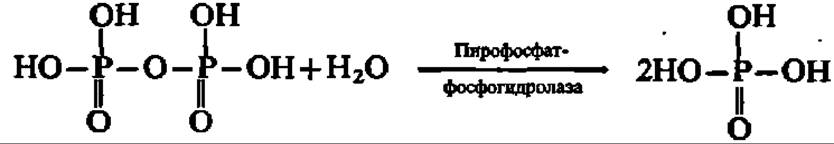
Две молекулы фарнезилпирофосфата соединяются по месту присоединения пирофосфатных группировок с отщеплением последних. Источником атомов водорода при образовании молекул пирофосфата является НАДФН. Уравнение данной реакции можно представить в виде следующей схемы:
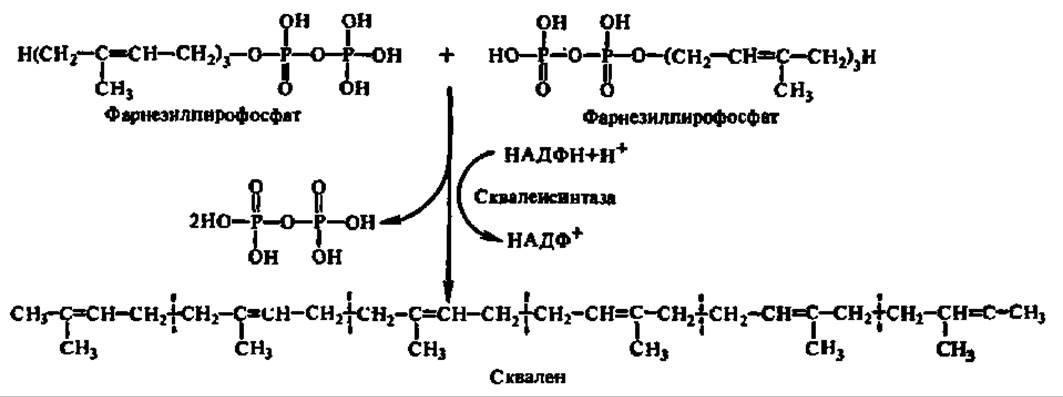
В результате реакции получается непредельный углеводород — сквален, составленный из 6 изопреноидных группировок. Процесс ускоряется сквален-синтазой, изучение которой затруднено ее тесной ассоциацией с эндоплазматической сетью и ее фосфолипидной составляющей. Однако выяснено, что при извлечении фермента из микросомальной фракции без применения детергентов (с использованием ультразвуковой дезинтеграции и т. п.) его М = 450 000, а с использованием детергента — 54500. Сквален-синтаза обладает двумя центрами связывания фарнезилпирофосфата, расположенными, возможно, на разных субъединицах.
Молекула сквалена легко принимает пространственную конфигурацию, близкую к пространственной конфигурации стеролов, и столь же легко окисляется по крайней двойной связи с образованием сквален-2,3-оксида с помощью сквален-эпоксидазы, относящейся к подклассу моноксигеназ (см. с. 419): Протонирование эпоксидной группы приводит к сдвигам электронной плотности в системе двойных связей сквалена и замыканию (показано стрелками) шестичленных и пятичленных циклов, характерных для стеролов. Схема указанного перехода скваленоксида в стерол представлена ниже:
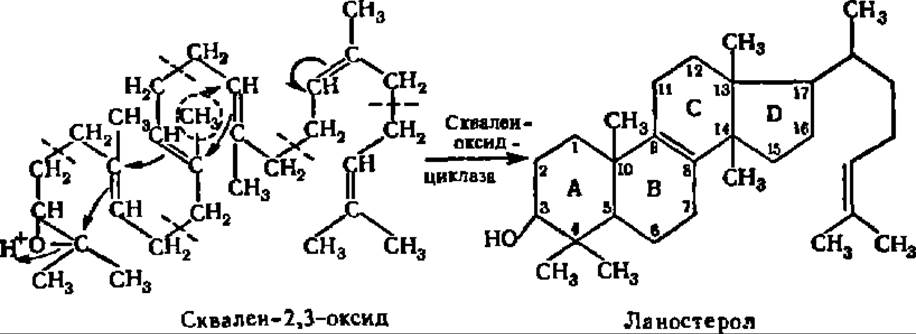
Ее особенность состоит в том, что при замыкании кольца С стерола неизбежно происходит переброска СН3-группы из положения 8 в положение 13 и элиминирование протона от 9-го углеродного атома цикла.
Так протекает процесс в эндоплазматической сети клеток печени. Что касается растений и других организмов, то циклизация сквален-2,3-оксида идет у них с помощью других циклизующих ферментных систем и с иными конечными продуктами, нежели ланостерол.
Путем преобразования ланостерола и других первичных продуктов циклизации возникают разнообразные индивидуальные стеролы, характерные для животного и растительного царства. Преобразование это многоступенчато; например, только удаление двух метильных групп при 4-м углеродном атоме кольца А (путем их окисления и последующего декарбоксилирования) осуществляется в 12 стадий.
Биосинтез стеридов протекает путем переноса остатка высшей жирной кислоты от молекулы ацил-КоА на место водорода ОН-группы стерола при каталитическом воздействии холестерол-ацилтрансферазы:
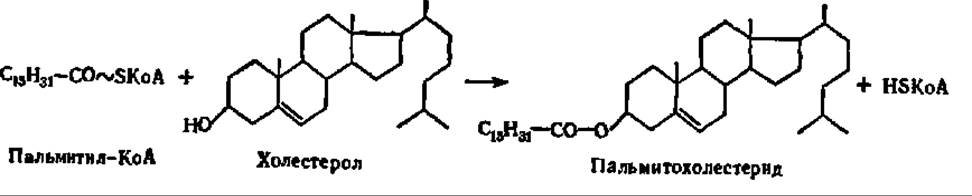
Источником ацильных групп при биосинтезе стеридов может также служить фосфатидилхолин. Так, например, синтезируются холестериды лимфы и плазмы крови у человека при участии фосфатидилхолин-холестерол-ацид-трансферазы.
В заключение подчеркнем, что диметилаллилпирофосфат и изопентенилпирофосфат служат универсальными исходными соединениями для биосинтеза ряда других полиизопреноидов — каротиноидов, каучука и т. п.
Обмен фосфатидов. Пути распада фосфатидов. Современные представления о путях распада фосфатидов в организме основаны главным образом на тщательном изучении превращений, которые свойственны фосфатидам вне организма при воздействии на них теми или иными ферментами. Поэтому когда говорят о путях распада фосфатидов, то имеют в виду скорее возможные, чем действительные, пути их деструкции. Непосредственно в биологических объектах эти пути исследованы еще недостаточно. Однако известно, что время полужизни фосфатидилглицерина и дифосфатидилглицерина у бактерий составляет 1 и 2 ч соответственно, а период полужизни фосфоинозитидов и сфингомиелинов в мозге крысы — 12,5 и 40 суток соответственно.
Фосфатиды распадаются на составляющие их структурные единицы — высшие жирные кислоты, фосфорную кислоту, азотистые основания и глицерин — гидролитическим путем. Реакции гидролиза, приводящие к разрушению сложноэфирных связей в молекуле фосфатида, ускоряются ферментами — фосфолипазами, которые относятся к подклассу эстераз (класс гидролаз). В зависимости от того, гидролиз какой из четырех имеющихся в молекуле фосфатида сложноэфирных связей ускоряет фосфолипаза, ее характеризуют как фосфолипазу А, В, С или D (схема 11).
Из схемы видно, что фосфолипазы A1 и А2 ускоряют реакцию отщепления а- и ß-ацильных радикалов в молекуле фосфатида; они характерны для животных и локализованы первая в эндоплазматической сети, а вторая — в митохондриях, образуя при гидролизе ß-ацил-лизолецитин и а-ацил-лизолецитин соответственно. Фосфолипазы А могут секретироваться и присутствуют, например, в ядах змей. У животных есть также фосфолипаза В, действующая на обе связи. Путь распада фосфатидов, открывающийся действием фосфолипазы С, присущ микроорганизмам, а фосфолипазы D — растениям.
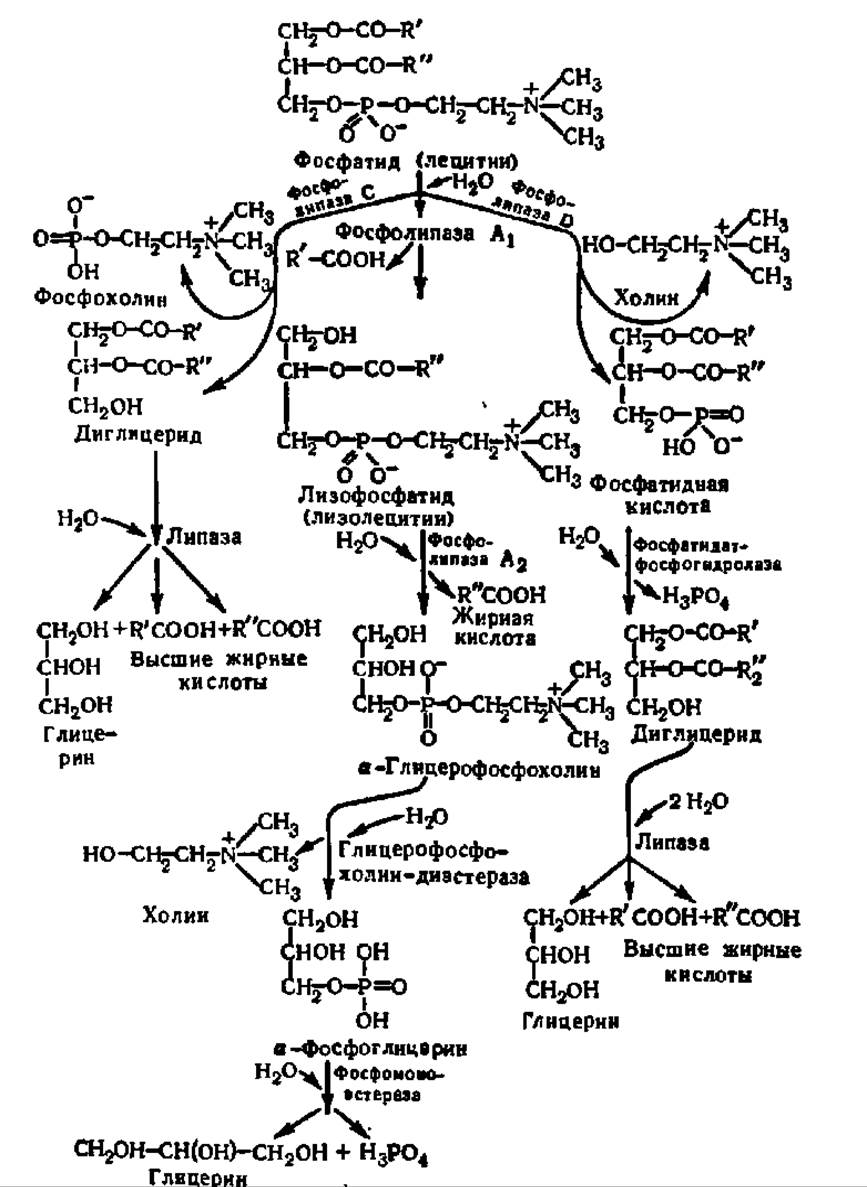
Схема 11. Пути распада фосфолипидов
Наиболее детально изучены, включая первичные и вторичные (рис. 127, А) структуры, фосфолипазы А2 из ядов змей и других источников. При молекулярных массах в 11 000—15 000 и минимум 4 дисульфидных мостиках они имеют активный центр, содержащий радикалы гистидина и аспарагиновой кислоты и работающий по схеме, характерной для ускорения гидролитических реакций (см. с. 332). Установлено также, что фосфолипазы функционируют в виде димеров, где одна субъединица осуществляет каталитический акт, а вторая — удаление отщепленного остатка высшей жирной кислоты (рис. 127, Б).
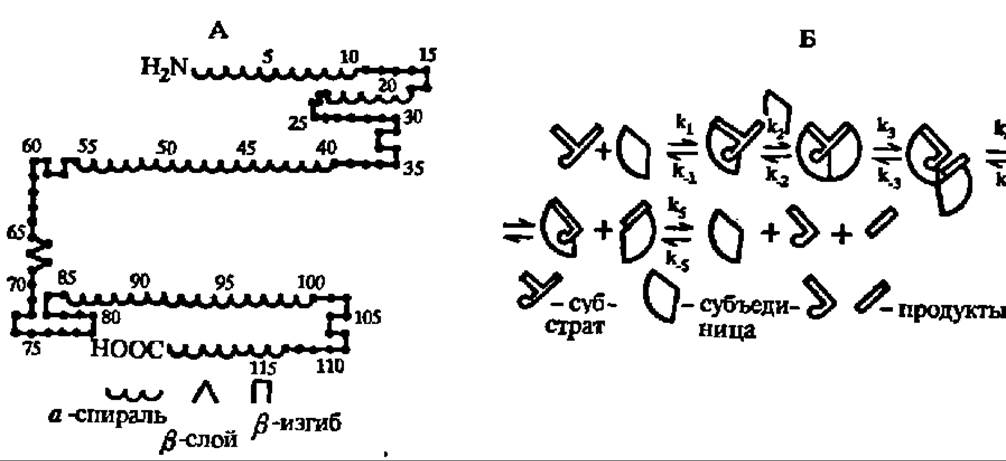
Рис. 127. Гипотетическая схема гидролиза фосфатидов фосфолипазой А2 из яда среднеазиатской кобры:
А — вторичная структура фосфолипазы А2; С — схема гидролиза — на 1-й ступени реакции (k3) возникает фермент-субстратный комплекс, в котором ацильная группа при а-углеродном атоме остатка глицерина располагается в субстратном центре, а остаток фосфорной кислоты с присоединенным к нему азотистым основанием — в каталитическом центре фосфолипазы А2. На 2-й ступени (k2) к фермент-субстратному комплексу присоединяется вторая молекула фермента, связывающая в своем субстратном центре ацильный радикал при ß-углеродном атоме оствтка глицерина молекулы фосфатида. На 3-й ступени реакции (k3) осуществляется гидролиз сложноэфирной связи при ß-углеродном атоме и удаление из зоны реакции ацильного радикала высшей жирной кислоты. На 4-й ступени (k4) димер фосфолипазы А2 распадается: с одним из протомеров остается связанным а-лизофосфатид, с другим — высшая жирная кислота. На 5-й ступени (k5) высвобождаются конечные продукты реакции
Особо следует подчеркнуть, что действие фосфолипаз на мембраны субклеточных частиц несомненно приводит к существенным сдвигам в функциональной активности последних. Этой роли фосфолипаз в обмене веществ и его регуляции придают в последнее время все большее значение.
Дальнейший обмен продуктов распада фосфатидов — высших жирных кислот и глицерина, был освещен ранее. Поэтому рассмотрим здесь лишь последующие превращения холина.
Одной из важнейших реакций, в которую вступает холин, по крайней мере, в нервной ткани животных, является реакция его ацетилирования. Источником ацетильной группы при этом служит ацетил-КоА, сама реакция ускоряется специфическим ферментом — холин-ацетилтраисферазой:
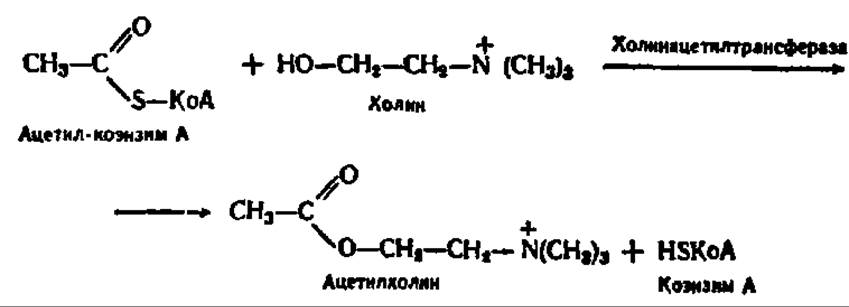
Ацетилхолин физиологически активен, так как он участвует в передаче нервных импульсов. Именно поэтому, видимо, фосфатиды, в частности холинфосфатиды. являются непременной составной частью нервной ткани.
Другой реакцией, имеющей существенное значение в обмене веществ, считают реакцию окисления холина в бетаин, который, в свою очередь, служит отличным донором СН3-групп в реакциях трансметилирования (см. с. 170):
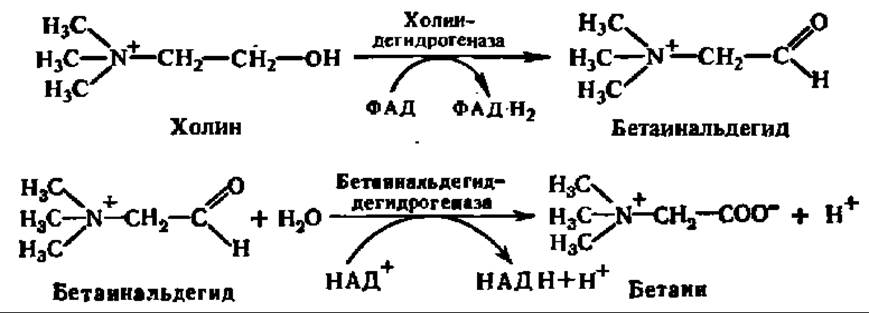
Бетаин, вступая в реакцию трансметилирования с гомоцистеином, образует метионин:
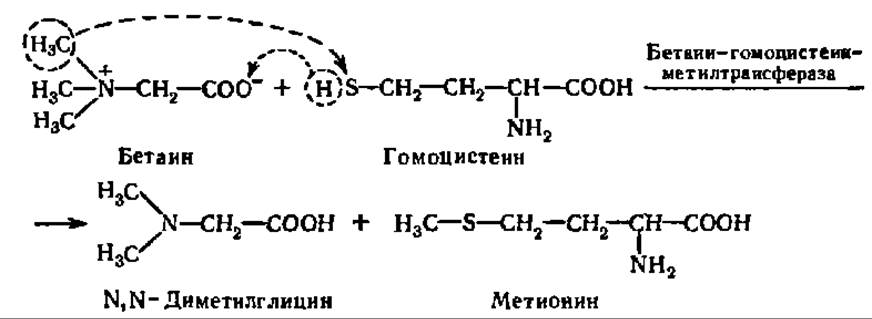
Как было указано выше, метионин в виде S-аденозилметионина является универсальным источником метальных групп в реакциях трансметилирования (см. с. 270).
Не исключено, что диметилглицин теряет оставшиеся две метальные группы при атоме азота и превращается в глицин.
Механизм биосинтеза фосфатидов. Как и во многих ранее отмеченных случаях, биосинтез фосфатидов протекает совсем иным путем, чем обращение реакций их гидролиза. Первые стадии биосинтеза фосфатидов совпадают с таковыми синтеза триглицеридов. Все идет одинаково вплоть до образования фосфатидной кислоты, а из нее — диглицерида. Однако дальше в случае биосинтеза фосфатидов на свободную ОН-группу диглицерида присоединяется остаток фосфохолина, который переносится из состава цитидиндифосфат- холина (ЦДФ-холин) (см. верхнее уравнение реакции на с. 409).
Указанный путь биосинтеза фосфатидов был открыт Е. Кеннеди и С. Вейсом (1956). Реакция ускоряется специфическим ферментом — ЦДФ-холин-1,2-диацилглицеролхолинфосфотрансферазой, локализованной в цитозоле (М = 200 000) и олигомеризующейся в присутствии диацилглицеринов с семикратным возрастанием активности, которая таким образом саморегулируется. Аналогично этому идет перенос остатка фосфоэтаноламина с ЦДФ-этанол-амина на диглицерид при участии также специфического фермента. Следовательно, такой путь биосинтеза вполне обоснован для лецитинов (холинфосфатидов) и кефалинов (коламинфосфатидов).
Возникает вопрос, каким же образом синтезируется в организме столь сложное соединение, как ЦДФ-холин? Механизм его биосинтеза таков (см. нижнее уравнение реакции на с. 409).
ЦМФ, взаимодействуя с АТФ, переходит снова в ЦТФ и, соединяясь с фосфохолином, опять образует ЦДФ-холин. Следовательно, ЦДФ-холин выполняет в этом процессе каталитическую функцию, перенося остатки фосфохолина на диглицерид. Если сравнить этот процесс с синтезом олиго- и полисахаридов, то там подобную функцию выполняла УДФ-глюкоза в отношении остатка глюкозы. Это дает основание утверждать, что в реакциях биосинтеза соединения типа нуклеозиддифосфатрадикалов играют выдающуюся роль как доноры тех или иных остатков органических соединений. Возможно, эта роль нуклеозиддифосфатпроизводных связана с их способностью превращать стабильную энергию макроэргической связи между остатками фосфорной кислоты в подвижную энергию возбуждения электронов взаимодействующих молекул, что и обеспечивает протекание реакции.
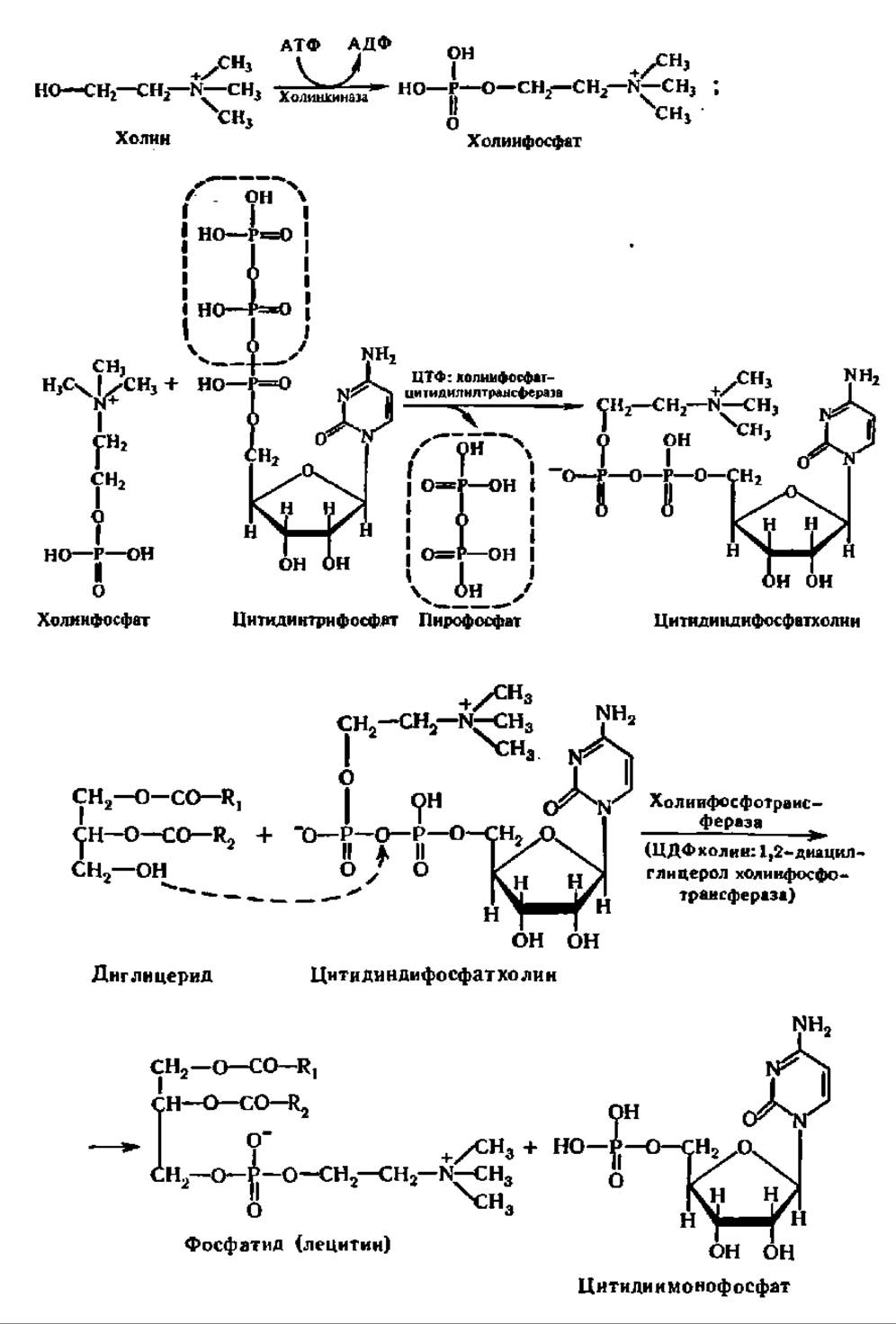
Ферменты и промежуточные продукты приведенного выше цикла реакций обнаружены в большинстве тканей животных, печени птиц, моркови и т. д. Особенно ярко они представлены в мозге. Впрочем, у дрожжей, например, синтез фосфатидилхолина идет главным образом за счет метилирования фосфатидилэтаноламина; это — второй путь биосинтеза лецитина, открытый Дж. Бретеном и Г. Гринбергом (1960). Фосфатидилглицерин, дифосфатидил-глицерин, фосфатидилинозит и фосфатидилсерин синтезируются через цитидин- дифосфатидилглицерины, возникающие из ЦТФ и диглицеридов под воздействием ЦДФ-диглицеридсинтазы (М = 114 000, димер). Взаимодействуя с глицеролфосфатом, инозитом и серином, ЦДФ-диглицериды образуют перечисленные выше фосфатиды при участии соответствующих ферментов.
Что касается обмена некоторых других видов липидов (сфинголипиды, гликолипиды и т. п.), то он осуществляется в соответствии с теми принципами, которые были отмечены при рассмотрении реакций обмена триглицеридов и фосфатидов. Распад сфинголипидов, гликолипидов и т. п. осуществляется при участии гидролаз, а дальнейший обмен продуктов их гидролиза — путем типовых реакций деструкции соединений соответствующих классов: углеводов, высших жирных кислот и др., рассмотренных ранее. Биосинтез сфинголипидов и гликолипидов протекает при широком участии разнообразных ацил- и гликозилтрансфераз.
Перенос липидов между мембранами. В последние годы наметилось новое направление в изучении обмена липидов. Оно касается достаточно энергично протекающего процесса межмембранного переноса липидов, особенно фосфолипидов, из митохондрий в эндоплазматическую сеть и обратно, из мембранной фракции клетки в липосомы, от липосом одного состава к липосомам другого состава, от внутреннего липидного слоя мембраны к внешнему и наоборот и т. п. Значение этого динамично протекающего обновления и видоизменения липидного состава мембран огромно, так как при его посредстве регулируется метаболическая активность мембранного аппарата клетки и субклеточных структур.
Важно, что межмембранный перенос липидов осуществляется специфическими белками, имеющими повсеместное распространение. Так, например, из цитозоля клеток печени быка выделен белок, переносящий фосфатидилхолин от одних мембран к другим. Молекулярная масса этого белка, связывающего и переносящего одну молекулу фосфатидилхолина, равна 22000, рІ = 5,8, в нем 190 аминокислотных остатков, 38% которых полярны.