Химия и биология белков - Ф. Гауровитц 1953
Белки с ферментативными свойствами
Механизм ферментативного гидролиза
Второй фазой ферментативного гидролиза является гидролитический распад субстрата AB на АОН и ВН. Поскольку в отсутствие фермента не происходит в заметной степени гидролитического распада AB, следует предположить, что распаду подвергается не AB, а комплекс Е(АВ). В этом комплексе связи между А и В расшатаны, что облегчает протекание гидролитического распада. Мы еще не знаем, каким путем происходит расшатывание связей между А и В. Наиболее вероятным представляется предположение о том, что в результате соединения молекулы субстрата с ферментом происходит ее деформация и либо под влиянием механических, сил, либо под влиянием электростатических воздействий соседних полярных групп наступает разрыв связей AB. Такие молекулы называют активированными.
Путем доставки энергии извне (например, тепловой энергии) можно молекулы AB перевести в активированное состояние AB*. Энергия, необходимая для перевода молекул в активированное состояние, носит название энергии активации. В гл. VII указывалось что энергия активации ∆Н* равна ∆F* + Т∆S*, где ∆F* — свободная энергия активации, Т — абсолютная температура и ∆S* — изменение энтропии. Если предположить, что распад фермент-субстратного комплекса на свободный фермент и продукты реакции происходит очень быстро, то скорость каталитической реакции будет зависеть главным образом от скорости процесса активации. В свою очередь, скорость процесса активации будет зависеть от величины свободной энергии активации ∆F* (см. стр. 164). Свободная энергия активации, требуемая для гидролиза сложных эфиров водородными ионами, равна примерно 10 000—13 000 кал/моль; для гидролиза пептидных связей — примерно 20 000 кал/моль. При ферментативных реакциях энергия активации уменьшается примерно до 4 000 кал/моль для гидролиза эфиров и до 12 000— 14 000 кал/моль для гидролиза пептидов [41]. Это действие фермента может быть выражено следующими уравнениями:
![]()
С термодинамической точки зрения ферменты и другие катализаторы можно определить как вещества, увеличивающие скорость химических реакций или обусловливающие возможность протекания этих реакций путем снижения величины свободной энергии активации. Как и все положения термодинамики, это определение затрагивает только энергетический баланс реакций и оценивает возможность протекания этих реакций с термодинамической точки зрения; оно, однако, не дает никакого представления о самом механизме ферментативных реакций, т. е. о природе реагирующих групп фермента и субстрата и о характере промежуточных соединений.
Для того чтобы получить некоторые представления о механизме ферментативных реакций, необходимо обратиться к изучению так называемых ферментных моделей, т. е. наиболее простых веществ, обладающих сходной с ферментом каталитической активностью.
До недавнего времени самыми простыми катализаторами процесса гидролиза считались сильные кислоты и основания. Действие этих веществ сводили к гидролизующему действию высокой концентрации ионов водорода или гидроксила. Большинство гидролитических ферментов имеет оптимум pH около 7, т. е. при такой реакции, при которой концентрация водородных и гидроксильных ионов очень мала. Это свидетельствует о том, что механизм ферментативного гидролиза, повидимому, отличен от механизма гидролиза сильными кислотами или основаниями. В последние годы, однако, была открыта новая группа гидролитических катализаторов. Штейнгардт [42] обнаружил, что различные органические сульфоновые кислоты, например додецилсульфонат или оранжевый II, обладают способностью гидролизовать амидные группы даже при низких концентрациях сульфоновой кислоты:
RCONH2→ RCOOH + NH3.
Сходным образом и белки подвергаются гидролизу под действием 0,1—0,01 М растворов додецилсульфоната при 65°, тогда как для гидролиза белков соляной или серной кислотами требуется обычно значительно более высокая концентрация [43]. Следует отметить, что гидролиз белков сульфоновыми кислотами не доходит до конца и останавливается при отщеплении приблизительно 50% аминокислот [43].
Каталитической активностью обладают не только растворимые в воде сульфоновые кислоты, но также и нерастворимые смолы, содержащие группы сульфоновой кислоты. Так, например, амберлит IR 100 (смола, содержащая 1 гэкв SО3H в 500 г вещества) гидролизует при 25° уксусноэтиловый эфир [44], а смола, представляющая собой производное фенолформальдегидсульфоновой кислоты, катализирует синтез сложного эфира из олеиновой кислоты и бутилового спирта [45]. Очевидно, группа сульфоновой кислоты в органических соединениях имеет большую каталитическую активность, чем серная кислота той же концентрации. Поскольку постоянные диссоциации органических кислот ниже постоянной диссоциации серной кислоты, нет оснований предполагать, что каталитическая активность сульфоновых кислот обусловлена большей по сравнению с серной кислотой концентрацией водородных ионов. Активность сульфоновых кислот зависит, повидимому, от структуры органической части молекулы сульфоновой кислоты. Можно предположить, что органический скелет как бы играет роль апофермента, тогда как группа сульфоновой кислоты выполняет функции кофермента.
Всем приведенным выше наблюдениям можно, однако, дать и совершенно иное толкование, а именно: можно предположить, что каталитическая активность фермента зависит от скопления большого количества ионных групп на ограниченном пространстве белковой молекулы фермента. Хорошо известно (хотя это явление до сих пор остается необъясненным), что активность ионов в концентрированных растворах щелочей или минеральных солей более чем в 100 раз выше теоретической величины. По аналогии можно представить, что скопление большого количества полярных групп в какой-нибудь небольшой части белковой молекулы приведет к необычайно высокой активности и, следовательно, к высокой каталитической эффективности. Действительно, было найдено, что четырехвалентные ионы лантана способны катализировать гидролиз мета- или пирофосфатов с образованием ортофосфата [46]. Выше уже указывалось, что многие пептидазы [16] и некоторые фосфатазы [17] содержат ионы металлов, например Mg++ или Мn++. В фермент-субстратном ком
плексе связь между полярными группами фермента и полярными группами пептида осуществляется через ионы металла. При такой форме связи могут быть получены кольца, в которых электронная структура пептидной связи окажется расшатанной до такой степени, что начнется процесс гидролиза [15, 16, 26, 47].
Механизм образования фермент-субстратного комплекса удобно проследить на реакциях, катализируемых фосфомутазами или киназами. Одним из этих ферментов является фосфоглюкомутаза, катализирующая равновесие между глюкозо-1-фосфатом и глюкозо-6-фосфатом. Фермент содержит прочно связанный фосфор, который нельзя удалить диализом. Однако при добавлении к ферменту глюкозо-1-фосфата, содержащего радиоактивный фосфор, происходит обмен атомами фосфора между ферментом и глюкозо-1-фосфатом [48]. Эта реакция в схематической форме может быть изображена следующим образом [49, 50]1:
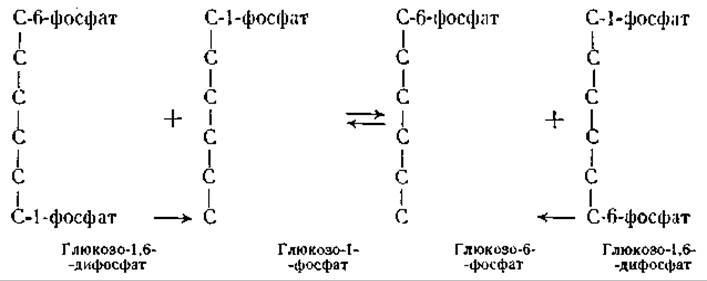
Как видно из приведенных формул, фосфатный остаток переносится ферментом с первого углеродного атома глюкозо-1,6-дифосфата на шестой углеродный атом глюкозо-1-фосфата и с шестого углеродного атома дифосфата на первый углеродный атом глюкозо-6-фосфата. Это доказано при помощи глюкозофосфатов, меченных С14. Очевидно, реакция может происходить только в присутствии глюкозо-1,6-дифосфата; поскольку дифосфат непрерывно регенерируется, для реакции требуются только следы этого вещества [49, 50]. Сходные результаты были получены с фосфоглицеромутазой, катализирующей взаимное превращение 2-фосфоглицериновой кислоты в 3-фосфоглицериновую кислоту [50]. В обоих случаях действие фермента сводится к тому, что он, временно соединяясь с остатком фосфорной кислоты, переносит его с одной молекулы на другую. Подобным же образом, как фосфоферазы, действуют миокиназа и гексокиназа (см. ниже). Если фосфатный остаток переносится на молекулу воды, то происходит гидролиз органического фосфорсодержащего соединения. В соответствии с этим фосфатазы можно рассматривать как фосфоферазы, катализирующие перенос фосфатного остатка с органических соединений на молекулу воды или с неорганического фосфата на органические соединения. Сходным образом и протеолитические ферменты можно рассматривать как аминоацил- феразы, которые, соединяясь с аминокислотным остатком, переносят его на другую аминокислоту, пептид или на молекулу воды. Если происходит перенос аминокислотного остатка на другую аминокислоту, то образуются новые пептиды, при перенесении же аминокислотного остатка на молекулу воды происходит гидролиз с освобождением аминокислот.
1 В английском издании книги автором при изложении процесса переноса фосфатных остатков с глюкозо-1,6-дифосфата на глюкозо-1-фосфат и глюкозофосфат были допущены ошибки как в формулах, изображающих этот процесс, так и в последующем пояснительном тексте. При переводе книги эти ошибки были исправлены, и схема переноса фосфатных остатков н пояснительный текст приводятся в том виде, в каком они даны в оригинальной работе Е. W. Sutherland, М. Cohn, Т. Posternak and С. Соmі (J. Biol. Chem., 180, 1285, 1949), на которую ссылается автор данной книги. — Прим. Ред.
Из всего сказанного следует, что соединение фермента со своим субстратом зависит главным образом от конфигурации и пространственного расположения реактивных групп фермента и субстрата. Если реактивная группа фермента может прийти в тесное соприкосновение с реактивной группой субстрата, то образуется фермент-субстратный комплекс и происходит каталитическая реакция.
До настоящего времени о реактивных группировках гидролаз известно весьма немногое. Некоторые данные относительно реактивных группировок их субстратов можно получить, изучая скорости каталитических реакций в присутствии конкурентных ингибиторов [51]. Конкурентными ингибиторами будут все вещества, близкие к субстратам гидролитических ферментов в структурном отношении, но отличающиеся от них некоторыми особенностями молекулярной структуры. Обладая, как и субстрат, способностью соединяться с ферментом, они конкурируют с ним и тем самым снижают скорость каталитической реакции.
Такого рода опыты показали, что ароматическое кольцо тирозина или фенилаланина имеет существенное значение для действия химотрипсина или карбоксипептидазы на полипептиды [20, 52]. Очевидно, в реакции соединения химотрипсина или карбоксипептидазы со своими субстратами участвует электронная система ароматического кольца. Поскольку бензольное кольцо не ионизировано и не является диполем, нет оснований полагать, что оно соединяется с полярными группами фермента. Возможно, что ароматическое кольцо субстрата соединяемся с подобным по строению ароматическим кольцом фермента.
Из изложенного видно, что хотя многие детали механизма образования фермент-субстратного комплекса еще не ясны, однако несомненно, что в его образовании участвуют несколько реактивных групп субстрата и несколько реактивных групп фермента. Это находится в соответствии с данными о высокой специфичности ферментативных реакций и подтверждает мнение о том, что формы поверхностей реагирующих групп фермента и субстрата дополняют друг друга.