ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 1. ОСНОВЫ БИОХИМИИ СТРОЕНИЕ И КАТАЛИЗ - 2011
ЧАСТЬ I. СТРОЕНИЕ И КАТАЛИЗ
3. АМИНОКИСЛОТЫ, ПЕПТИДЫ И БЕЛКИ
3.3. Как работать с белками
Наши представления о структуре и функции белка основаны на результатах исследований множества различных белков. Для проведения таких исследований биохимик должен уметь отделить свой белок от других и владеть методами, позволяющими изучать его свойства. Совокупность таких экспериментальных методов и составляет суть предмета химии белка — дисциплины старой, как сама биохимия, и занимающей одно из центральных мест в биохимических исследованиях.
Белки можно разделить и очистить
Прежде чем приступить к изучению свойств белка, необходимо получить его чистый препарат. Как это сделать, если клетки содержат тысячи различных типов белков? Методы разделения основаны на том, что все белки различаются по размерам, заряду и способности связываться с другими веществами. Некоторые другие современные методы, включая клонирование ДНК и секвенирование геномов, могут упростить проведение очистки белка (см. гл. 9).
Обычно источником белка служат ткани или микробные клетки. Первой стадией любого метода выделения белка является разрушение клеток, в результате которого содержащиеся в них белки переходят в раствор, называемый грубым экстрактом. Для извлечения отдельных клеточных фракций или специфических органелл иногда на этом этапе применяют дифференциальное центрифугирование (рис. 1-8).
Существует множество методов, позволяющих выделить один или несколько белков из грубого экстракта или препарата органелл. Обычно экстракт подвергают процедуре, позволяющей разделить белки на фракции, пользуясь различиями в размерах или зарядах белковых молекул. Данный процесс называется фракционированием. На первых стадиях фракционирования пользуются различной растворимостью белков, зависящей от таких условий, как pH, температура, концентрация соли и др. Растворимость белков обычно снижается при повышении концентрации соли в растворе; данный эффект называют «высаливанием» или осаждением солями. Добавление определенной порции соли может вызвать селективное осаждение некоторой фракции белков, в то время как другие все еще будут оставаться в растворе. Сульфат аммония (NН4)2SO4 очень часто используется для высаливания белков. С помощью этой процедуры некоторые белки переводят в осадок (который затем удаляют путем центрифугирования на низкой скорости) и отделяют их тем самым от других белков, остающихся в растворе.
Далее раствор белка можно подвергнуть диализу — процедуре, при которой белки отделяются от других растворенных веществ благодаря своему большому размеру. Частично очищенный экстракт помещают в мешочек из полупроницаемой мембраны и погружают в большой объем буфера с определенной ионной силой; через мембрану могут проходить соли и компоненты буфера, но не белки. В результате диализа в мешочке остаются крупные белки, а концентрация солей в мешочке сравнивается с концентрацией в растворе за пределами мембраны. Диализ можно использовать, например, для удаления сульфата аммония из препарата белка.
Наиболее мощным методом фракционирования белков является колоночная хроматография, основанная на различии размеров, зарядов, аффинности и других свойств белков (рис. 3-16). Колонку заполняют твердым пористым материалом (носитель, или стационарная фаза), сквозь который просачивается буферный раствор (подвижная фаза). Раствор белка наносят на верхнюю часть колонки, и он начинает просачиваться сквозь твердую матрицу как постоянно расширяющаяся полоса внутри подвижной фазы. Отдельные белки движутся сквозь колонку медленнее или быстрее в зависимости от их свойств.
Рис. 3-16. Колоночная хроматография. Стандартная колоночная хроматография подразумевает наличие твердого пористого материала, помещенного в стеклянную или пластиковую колонку. Твердый материал (матрица, или носитель) создает стационарную фазу, сквозь которую протекает жидкость (подвижная фаза). Сверху из резервуара в колонку постоянно поступает раствор, он протекает сквозь колонку и выходит снизу (элюент). Раствор подлежащих разделению белков наносят в верхнюю часть колонки и дают ему войти в твердую фазу. Далее в колонку продолжают добавлять растворитель. Раствор белка образует внутри подвижной фазы полосу, ширина которой сначала равна слою внесенного образца. По мере прохождения через колонку белки начинают удерживаться на носителе за счет разнообразных взаимодействий. В результате общая полоса белка расширяется. Отдельные типы белков (А, В и С, показанные синим, красным и зеленым цветом) постепенно отделяются друг от друга, и каждый образует отдельную фракцию (полосу) внутри общей полосы белка. Чем больше длина колонки, тем лучше разделение, однако полоса каждого отдельного белка со временем также начинает расплываться за счет диффузии, что ухудшает разрешение. В данном примере белок А хорошо отделился от белков В и С, но из-за диффузии полное разделение В и С в данных условиях не представляется возможным.
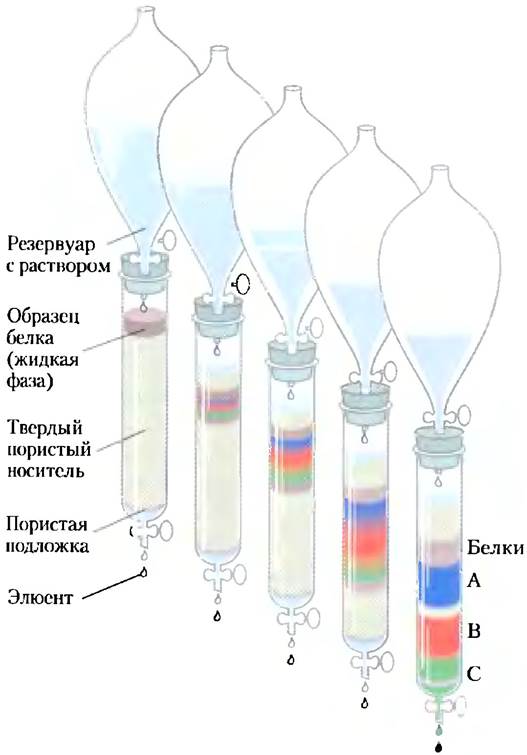
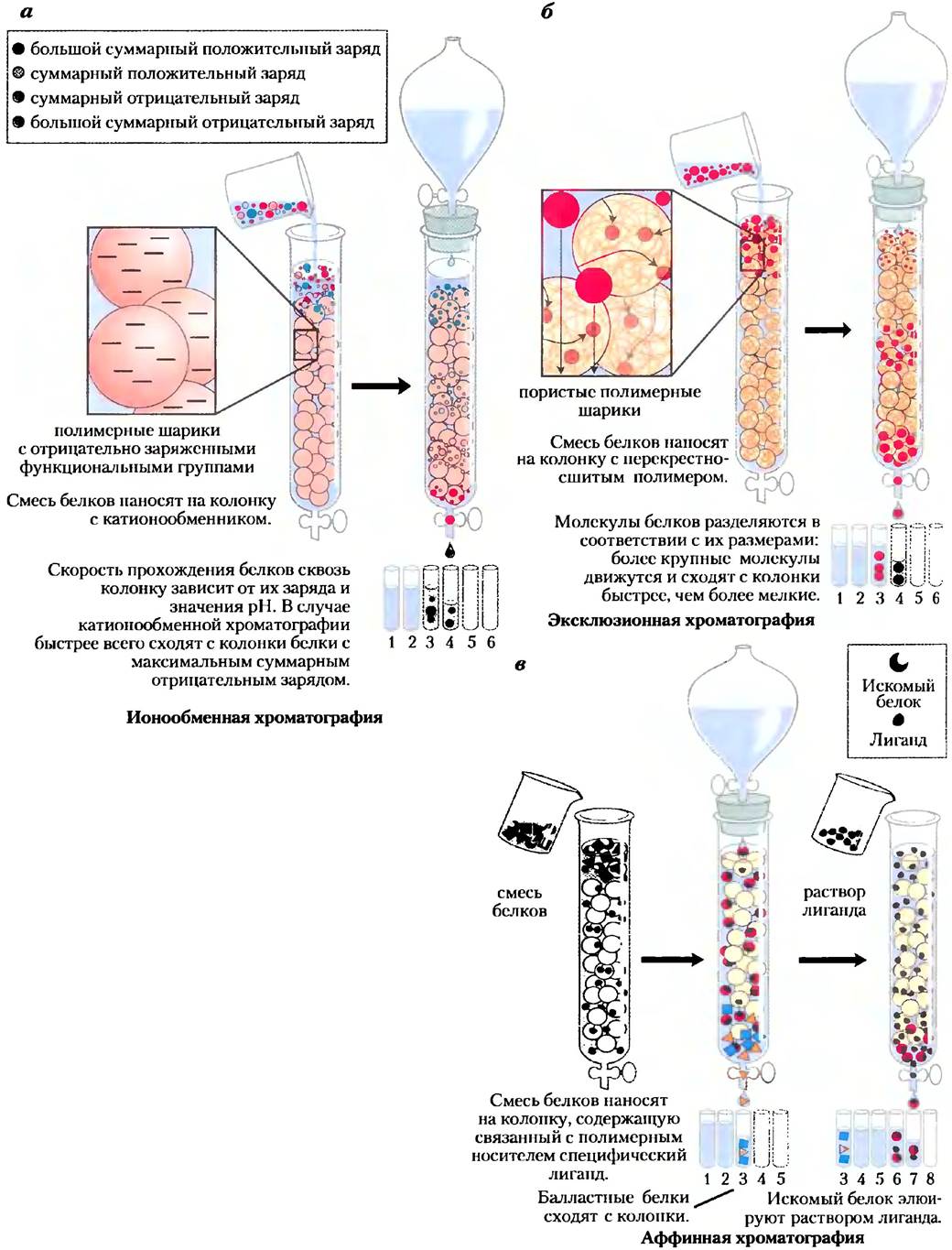
Ионообменная хроматография основана на различии знака и величины заряда белков при данном значении pH. Носитель, которым заполняют колонку, представляет собой синтетический полимер (смолу), содержащий связанные заряженные группы. Носители с анионными группами называют катионообменниками, а носители с катионными группами — анионообменниками. Сродство каждого белка к заряженным группам носителя зависит от pH (который определяет состояние ионизации молекулы) и концентрации конкурирующих свободных ионов в растворе. Улучшить разделение можно путем постепенного изменения pH и(или) концентрации солей в подвижной фазе, т. е. путем создания градиента pH или концентрации соли. При катионообменной хроматографии (рис. 3-17, а) стационарная фаза имеет отрицательно заряженные группы. Белок, несущий суммарный положительный заряд, движется в жидкой фазе медленнее, чем белок, несущий отрицательны заряд, поскольку продвижение первого белка тормозится за счет взаимодействия с носителем.
Рис. 3-17. Три хроматографических метода, применяемые при разделении белков, а) Ионообменная хроматография использует разницу в знаке и величине заряда белковых молекул при определенном значении pH. 6) Экск- люзионная хроматография (гель-фильтрация) позволяет разделить белки разного размера, в) Аффинная хроматография позволяет разделять белки в соответствии с их способностью связываться со специфическим носителем. Дальнейшее объяснение этих методов дано в тексте.
Расширение полосы белка в подвижной фазе объясняется как разделением белков с различными свойствами, так и диффузией. По мере увеличения длины колонки вероятность разделения двух белков с различными зарядами обычно возрастает, однако время разделения увеличивается. В результате разделение полос может даже ухудшиться, поскольку с увеличением длительности процесса преобладающий эффект оказывает диффузия. По мере выхода раствора с колонки фракции элюента собирают в отдельные пробирки. Затем каждую фракцию тестируют на присутствие искомого белка и других белков. Все фракции, в которых содержится искомый белок, можно объединить.
Пример 3-1. Разделение пептидов с помощью ионообменной хроматографии
Биохимик хочет разделить два пептида с помощью ионообменной хроматографии. При том значении pH, при котором происходит разделение на колонке, пептид А имеет суммарный заряд -3, поскольку в его последовательности чаще встречаются остатки Glu и Asp, чем Arg, Lys и His. Пептид В имеет суммарный заряд +1. Какой пептид будет первым сходить с колонки, заполненной катионообменной смолой? Какой пептид будет первым сходить с колонки, заполненной анионообменной смолой?
Решение. Катионообменная смола имеет на поверхности отрицательно заряженные группы и связывает положительно заряженные молекулы, замедляя их перемещение вдоль колонки. Пептид В, несущий положительный заряд, сильнее связывается с катионообменной смолой, чем пептид А, так что пептид А сходит с колонки первым. В случае анионообменной смолы первым будет выходить пептид В. Пептид А, несущий отрицательный заряд, задерживается за счет взаимодействия с положительно заряженными группами носителя.
На рис. 3-17, кроме того, представлены два других типа колоночной хроматографии. Эксклюзионная хроматография (также называемая ситовой хроматографией или гель-фильтрацией; рис. 3-17, б) позволяет разделить белки разного размера. В этом методе более крупные белки сходят с колонки быстрее мелких белков, что на первый взгляд противоречит интуитивным соображениям. Дело в том, что твердая фаза колонки состоит из шариков с порами или полостями определенного размера. Крупные белки не могут проникать в поры и поэтому просто проходят между шариками. Мелкие белки попадают в поры, задерживаются там, и в результате движутся сквозь колонку медленнее.
Аффинная хроматография основана на способности белков связываться с носителем (рис. 3-17, е). Шарики в колонке на своей поверхности несут присоединенные ковалентной связью химические группы, называемые лигандами; лиганд — это химическая группа или молекула, связывающаяся с макромолекулой (например, с белком). При нанесении на колонку смеси белков каждый белок, имеющий сродство к этому лиганду, связывается с шариками; в результате его продвижение вдоль колонки замедляется. Например, если биологическая функция белка подразумевает его связывание с АТР, то связывание АТР с шариками позволит создать аффинную матрицу, которую можно использовать для очистки этого белка. При прохождении раствора белка вдоль колонки все ATP-связывающие белки (включая искомый белок) связываются с матрицей. Когда все не связывающиеся с матрицей белки сходят с колонки, связавшиеся белки смывают раствором, содержащим либо высокую концентрацию соли, либо свободный лиганд (в данном случае АТР). Соль ослабляет связывание белка с иммобилизованным лигандом, нарушая ионные взаимодействия. Свободный лиганд конкурирует с иммобилизованным лигандом за связывание с белком, что приводит к снятию белка с матрицы. Однако в данном случае сходящий с колонки белок часто связан с лигандом, применявшимся для элюирования.
Наиболее усовершенствованным хроматографическим методом является высокоэффективная жидкостная хроматография (ВЭЖХ или HPLC, от англ. high-performance liquidchromatography). В данном методе для ускорения движения белковых молекул через колонку используют насосы, создающие высокое давление. Кроме того, применяют специально разработанные высококачественные хроматографические материалы, способные выдерживать условия высокого давления. Снижение времени прохождения белка через колонку позволяет ограничить диффузию и тем самым значительно улучшить разрешение.
Метод очистки нового белка каждый раз подбирают, исходя из опыта и здравого смысла. В большинстве случаев для очистки белка приходится последовательно применять несколько различных методов, основанных на разделении белков в соответствии с различными параметрами и свойствами. Например, если определенная стадия очистки позволяет отделить АТР-связывающие белки от белков, которые с АТР не связываются, то на следующей стадии очистки необходимо отделить искомый белок от остальных ATP-связывающих белков на основании различия размеров или зарядов. Выбор часто осуществляется эмпирически; иногда в поисках наиболее эффективного способа приходится опробовать множество разных подходов. Этот поиск иногда удается несколько сократить, если использовать методы очистки, применявшиеся ранее для аналогичных белков. На сегодняшний день опубликованы протоколы выделения тысяч самых разных белков. Здравый смысл подсказывает, что такие недорогие методы, как осаждение солями, следует использовать на начальных этапах очистки, когда общий объем смеси и количество примесей максимальны. Применение хроматографических методов на первых этапах непрактично, поскольку для разделения большого объема образца требуется большое количество дорогостоящих хроматографических сред. По мере очистки препарата его объем обычно сокращается (табл. 3-5), что делает возможным применение более сложных (и дорогих) хроматографических методов.
Таблица 3-5. Схема очистки гипотетического фермента
Метод очистки |
Объем фракции (ма) |
Общее количество белка (мг) |
Активность (единицы) |
Удельная активность (единины/мг) |
1. Грубый экстракт |
1 400 |
10 000 |
100 000 |
10 |
2. Осаждение сульфатом аммония |
280 |
3 000 |
96 000 |
32 |
3. Ионообменная хроматография |
90 |
400 |
80 000 |
200 |
4. Гель-фильтрация |
80 |
100 |
60 000 |
600 |
5. Аффинная хроматографии |
6 |
3 |
45 000 |
15 000 |
Замечание: численные данные отражают состояние образца после проведенной процедуры очистки. Определение активности и удельной активности приводится ниже (с. 140).
Белки можно разделить и охарактеризовать методом электрофореза
Еще один важный метод разделения белков основан на способности заряженных белков двигаться в электрическом поле — этот процесс называют электрофорезом.Электрофорез редко используют для очистки больших количеств белков, поскольку обычно существуют другие, более простые методы, кроме того, этот процесс часто нарушает структуру и биологическую активность белка. Однако электрофорез чрезвычайно широко используется как аналитический метод. Достоинство метода состоит в том, что белки можно не только разделить, но и визуализировать, что позволяет быстро оцепить количество белков в смеси или степень чистоты данного белкового препарата. Методом электрофореза можно также определить важнейшие характеристики белка, например, изоэлектрическую точку и молекулярную массу.
Электрофорез белков обычно проводят в геле, образованном перекрестно-сшитым полимерным веществом полиакриламидом (рис. 3-18). Полиакриламидный гель действует в качестве молекулярного сита, в котором скорость движения белков пропорциональна соотношению их заряда и массы. На скорость движения, кроме того, может влиять форма белковой молекулы. Силой, заставляющей двигаться молекулы в электрическом поле, является электрический потенциал (Е). Электрофоретическая подвижность молекулы (μ) представляет собой отношение скорости движения частицы (V) к электрическому потенциалу. Электрофоретическая подвижность, кроме того, равна отношению суммарного заряда молекулы (Z) к коэффициенту торможения (f), связанному с формой молекулы.
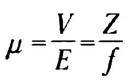
Таким образом, подвижность белка в геле в процессе электрофореза определяется его размером и формой.
Электрофорез, использующийся для оценки чистоты белка и его молекулярной массы, проводят в присутствии детергента додецилсульфата натрия (англ. sodium dodecylsulfate, SDS).
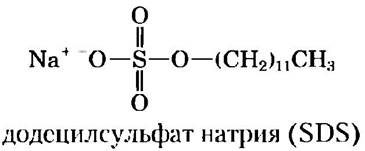
Додецилсульфат натрия связывается с большинством белков в количестве, пропорциональном молекулярной массе белка, приблизительно одна молекула SDS па каждые два аминокислотных остатка. В результате молекула белка приобретает большой отрицательный заряд, по сравнению с которым собственный заряд белка оказывается несущественным; кроме того, все белки в присутствии SDS характеризуются одинаковым отношением заряда к массе. При связывании молекул SDS белки частично разворачиваются и приобретают приблизительно одну и ту же форму. Таким образом, разделение белков методом электрофореза в присутствии SDS происходит почти исключительно в соответствии с их молекулярными массами: чем меньше белок, тем быстрее он движется. После проведения электрофореза белки визуализируют с помощью красителя Кумасси синего, который окрашивает белки, но не окрашивает сам гель (рис. 3-18,б).
Рис. 3-18. Электрофорез, а) В лунки в верхней части полиакриламидного геля вносят различные образцы белков. При подключении электрического тока белки начинают двигаться внутри геля. Гель минимизирует как конвекционные потоки, вызванные небольшими температурными перепадами, так и перемещение белка, за исключением того, что связано с движением в электрическом поле. б) После проведения электрофореза белки можно визуализировать, обработав гель раствором красителя Кумасси синего, который связывается только с белками, но не с гелем. Каждая полоса на геле соответствует отдельному белку (или субъединице белка); более мелкие белки мигрируют в геле быстрее, чем более крупные, и поэтому находятся в нижней части геля. Результаты очистки белка RecA из Escherichia coli (см. гл. 25). Ген белка RecA был клонирован (гл. 9), так что его экспрессию (синтез белка) можно контролировать. Первая дорожка представляет собой набор стандартных белков с известными значениями Mr которые служат в качестве маркеров. На двух следующих дорожках представлен набор белков E. coli до и после индукции синтеза RecA соответственно. На четвертой дорожке нанесены белки из грубого клеточного экстракта. Далее на дорожках (слева направо) последовательно представлены образцы после каждой стадии очистки. Очищенный белок имеет единственную полипептидную цепь с Mr = 38 000.
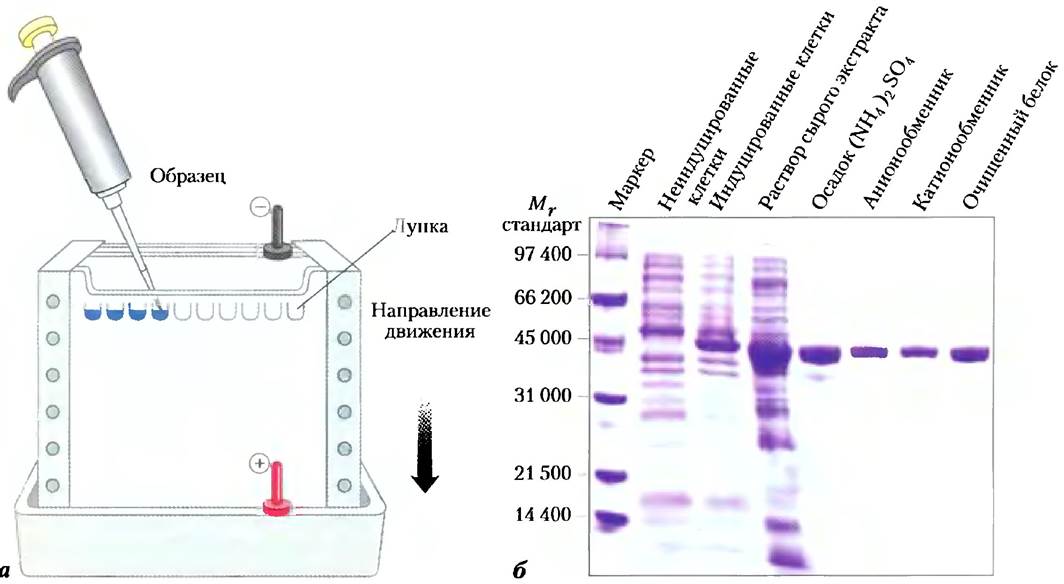
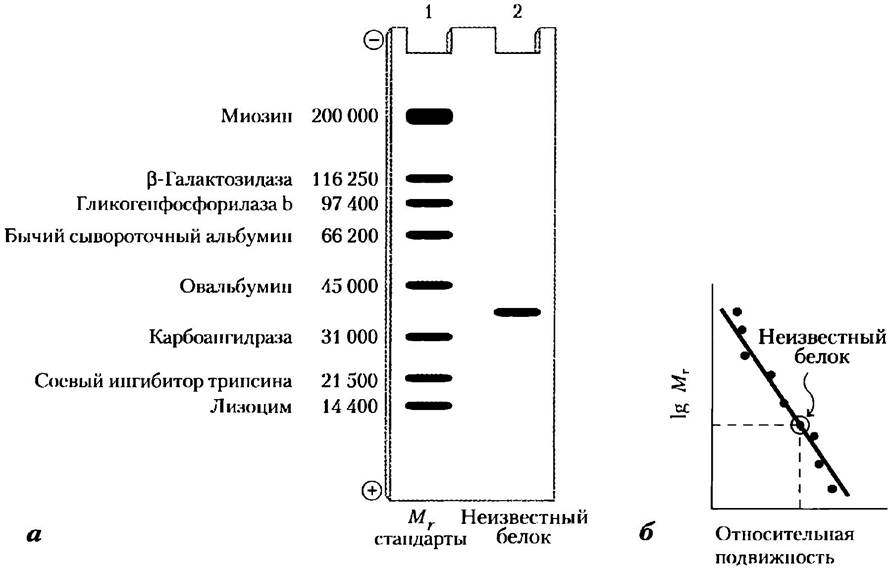
С помощью этого метода удобно следить за ходом очистки белка по количеству белковых полос, остающихся на геле после проведения каждой следующей стадии очистки. Сравнивая расстояния, пройденные в геле исследуемым белком и белками с известной молекулярной массой, можно оценить молекулярную массу исследуемого белка (рис. 3-19). Белки, состоящие из нескольких субъединиц, при проведении электрофореза в присутствии SDS обычно разделяются на отдельные субъединицы, каждая из которых видна как отдельная полоса. SDS гель- электрофорез.
Рис. 3-19. Оценка молекулярной массы белка. На основании подвижности белка в процессе SDS-электрофореза можно определить его молекулярную массу, а) Для определения неизвестной молекулярной массы белка (нанесен на дорожку 2) используют набор маркерных белков с известными молекулярными массами (дорожка 1). б) По графику зависимости относительной подвижности маркерных белков от логарифма их молекулярных масс можно найти значение молекулярной массы неизвестного белка.
Для нахождения численного значения изоэлектрической точки белка (рl) пользуются методом изоэлектрофокусирования (рис. 3-20). Градиент pH создается с помощью смеси низкомолекулярных органических кислот и оснований (амфолитов, с. 123), которые распределяются в геле под действием электрического поля. Если нанести на гель смесь белков, то в электрическом поле каждый из них движется до тех пор, пока не достигнет области pH, совпадающей с его значением pl (табл. 3-6). Таким образом, белки с разными значениями изоэлектрических точек локализуются в разных участках геля.
Рис. 3-20. Изоэлектрофокусирование. Данный метод позволяет разделить белки с различающимися значениями изоэлектрических точек. С помощью раствора подходящих амфолитов создают градиент pH в геле. Затем в лунку на геле наносят смесь белков. При подключении электрического поля белки входят в гель и движутся до тех пор, пока не достигнут тех областей, значения pH которых равны значениям изоэлектрических точек белков (поскольку при pH = рl суммарный заряд белка равен нулю).
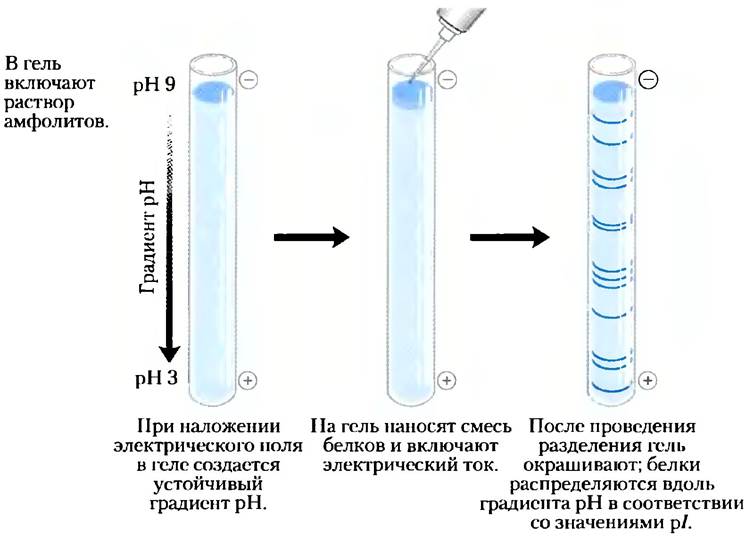
Таблица 3-6. Изоэлектрические точки некоторых белков
Белок |
pl |
Пепсин |
<1.0 |
Яичный альбумин |
4,0 |
Сывороточ пый альбумин |
4,9 |
Уреаза |
5,0 |
β-Лактоглобулин |
5,2 |
Гемоглобин |
0.8 |
Миоглобин |
7,0 |
Химотрипсиноген |
9.5 |
Циохром с |
10.7 |
Лизоцим |
11,0 |
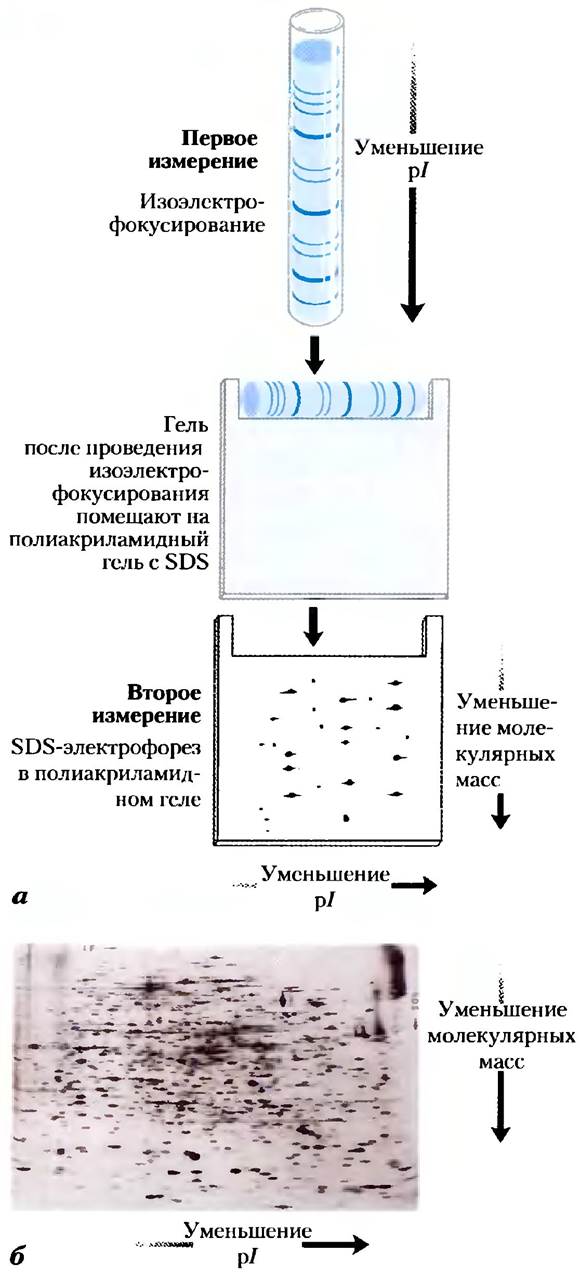
Сложные смеси белков можно разделить, если последовательно использовать изоэлектрофокусирование и SDS-электрофорез с помощью так называемого метода двумерного электрофореза (рис. 3-21). Этот метод более чувствителен, чем простой электрофорез. Двумерный электрофорез позволяет разделить белки с одинаковой молекулярной массой, но разными значениями рl, а также белки с одинаковым значением рl, но с разными молекулярными массами.
Рис. 3-21. Двумерный электрофорез, а) На первом этапе белки разделяют методом изоэлектрофокусирования в цилиндрическом геле. Затем гель помещают горизонтально на второй гель в виде пластины и подвергают белки разделению с помощью SDS-электрофореза. В горизонтальном направлении белки разделяются в соответствии со значениями рl, а в вертикальном направлении - в соответствии со значениями молекулярных масс. б) Таким способом удается разделить более 1000 белков, находящихся в клетках Е. coli.
Возможность контролировать содержание белка в неразделенных смесях
В процессе очистки белка необходимо иметь возможность контролировать его наличие и определять его количество в неразделенной смеси белков. Но часто приходится осуществлять очистку белка при полном отсутствии информации о его молекулярной массе, физических свойствах и о той доле общей массы белка, которую он составляет. Если речь идет о ферментах, то их количество в растворе или экстракте ткани можно измерить или хотя бы оценить на основании их каталитической активности, т. е. по увеличению скорости превращения субстрата в присутствии образца фермента. Для этого нужно знать: 1) общее уравнение ферментативной реакции; 2) аналитический метод, позволяющий измерять убыль субстрата или прирост продукта реакции; 3) зависимость активности фермента от каких-либо кофакторов (ионов металлов) или коферментов; 4) зависимость ферментативной активности от концентрации субстрата; 5) оптимум pH; 6) диапазон температуры, в котором фермент активен и стабилен. Анализ активности ферментов обычно проводят при оптимальных для них значениях pH и при температуре от 25 до 38 °С. Кроме того, обычно берут очень высокие концентрации субстратов, чтобы начальная скорость реакции была пропорциональна концентрации фермента (гл. 6).
Активность ферментов принято измерять в международных ферментативных единицах. В соответствии с международной договоренностью за одну единицу ферментативной активности принимают такое количество фермента, которое вызывает превращение 1 мкмоль субстрата за 1 мин при 25 °С при оптимальных условиях проведения реакции (для некоторых ферментов это определение некорректно, и их активность определяют по-другому). Под термином активность понимают общее количество единиц активности фермента в образце. Удельной активностью называют количество единиц ферментативной активности, приходящихся на 1 мг общего белка (рис. 3-22). Удельная активность является мерой чистоты фермента: она возрастает в процессе очистки, становится максимальной и больше не изменяется, когда фермент очищен окончательно (табл. 3-5, с. 136).
Рис. 3-22. Активность и удельная активность. Разницу между этими двумя понятиями можно объяснить на примере двух стаканов с шариками. В стаканах содержится одинаковое количество красных шариков, но различное количество шариков других цветов. Если под шариками подразумевать белки, то в стаканах содержится одинаковая активность белка, представленного красными шариками. Однако удельная активность этого белка во втором стакане выше, поскольку в нем красные шарики составляют более значительную долю от общего числа шариков.
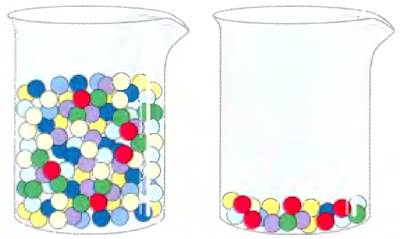
После каждой стадии очистки следует определять ферментативную активность образца (в единицах ферментативной активности) и общее количество белка; отношение этих двух значений дает удельную активность образца. Общая активность и общее количество белка обычно снижаются в процессе очистки препарата. Ферментативная активность снижается, поскольку очистка неминуемо сопряжена с некоторой инактивацией фермента, например, за счет неблагоприятных взаимодействий с хроматографическими средами или с другими молекулами. Общее количество белка снижается, поскольку целью очистки как раз и является удаление максимального количества ненужного или неспецифического белка. Стадию очистки можно считать удачной, если после ее проведения потери неспецифического белка значительно выше потерь активности; таким образом, удельная активность образца возрастает, несмотря на то что общая активность падает. Данные о стадиях очистки образца и их результатах объединяют в таблицы, аналогичные табл. 3-5. Белок считается чистым, если при проведении дальнейшей очистки не удастся повысить удельную активность, а также если с помощью аналитических методов (например, электрофореза) детектируется единственная белковая полоса.
Для белков, нс обладающих ферментативной активностью, необходимы другие методы обнаружения. Транспортные белки можно определять по связыванию с транспортируемыми ими молекулами, а гормоны и токсины — но оказываемому ими биологическому действию. Например, гормоны роста должны стимулировать рост определенных клеточных культур. Некоторые структурные белки представляют настолько значительную фракцию от общего белка клетки или ткани, что их удастся довольно легко выделить и очистить без проведения функционального анализа. Методов решения проблемы в этом смысле существует столько же, сколько и самих белков.
Краткое содержание раздела 3.3 Как работать с белками
■ Методы очистки и разделения белков основаны на различии их свойств. Белки можно селективно осадить, добавляя в раствор определенные соли. Существует несколько хроматографических методов разделения, основанных на разнице молекулярных масс, сродства, заряда и других свойств белков. Из хроматофафических методов чаще всего применяют ионообменную, эксклюзионную, аффинную и высокоэффективную жидкостную хроматографию.
■ Электрофоретические методы позволяют разделить белки с разной молекулярной массой или зарядом. Методы SDS-электрофореза и изоэлектрофокусирования можно использовать как по отдельности, так и в сочетании, что позволяет получить лучшее разрешение.
■ При осуществлении любой процедуры очистки белка необходим метод наблюдения за искомым белком на фоне всех других белков в смеси. Мерой очистки белка может быть его удельная активность.