ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 2. БИОЭНЕРГЕТИКА И МЕТАБОЛИЗМ - 2014
ЧАСТЬ II. БИОЭНЕРГЕТИКА И МЕТАБОЛИЗМ
18. ОКИСЛИТЕЛЬНОЕ РАСЩЕПЛЕНИЕ АМИНОКИСЛОТ И ОБРАЗОВАНИЕ МОЧЕВИНЫ
18.3. Пути деградации углеродного скелета аминокислот
В норме катаболизм всех аминокислот производит всего от 10% до 15% энергии в теле человека; он не такой активный как гликолиз и окисление жирных кислот. Различные потоки этого катаболического пути также сильно варьируют. Они зависят от баланса между требованиями процессов биосинтеза и доступностью, определенной аминокислоты. Двадцать путей распада аминокислот сходятся с образованием всего шести главных продуктов, и все они попадают в цикл трикарбоновых кислот (рис. 18-15). Отсюда углеродные скелеты поступают в глюконеогенез или кетогенез или полностью окисляются до СO2 и Н2O.
Рис. 18-15. Обзор катаболизма аминокислот. Аминокислоты сгруппированы в соответствии с главным продуктом их деградации. Некоторые аминокислоты встречаются более одного раза, так как участки их углеродных скелетов расщепляются до разных конечных продуктов. Показаны наиболее важные катаболические пути у позвоночных, но и между позвоночными существуют небольшие отличия. Треонин, например, распадается как минимум двумя разными путями (см. рис. 18-19,18-27), и важность определенного пути для организма может быть различной в зависимости от условий метаболизма. Глюкогенные и кетогенные аминокислоты выделены разными цветами. Обратите внимание на то, что пять аминокислот являются и глюкогенными, и кетогенными. Аминокислоты, разлагающиеся до пирувата, также потенциально кетогенные. Только две аминокислоты, лейцин и лизин, исключительно кетогенные.
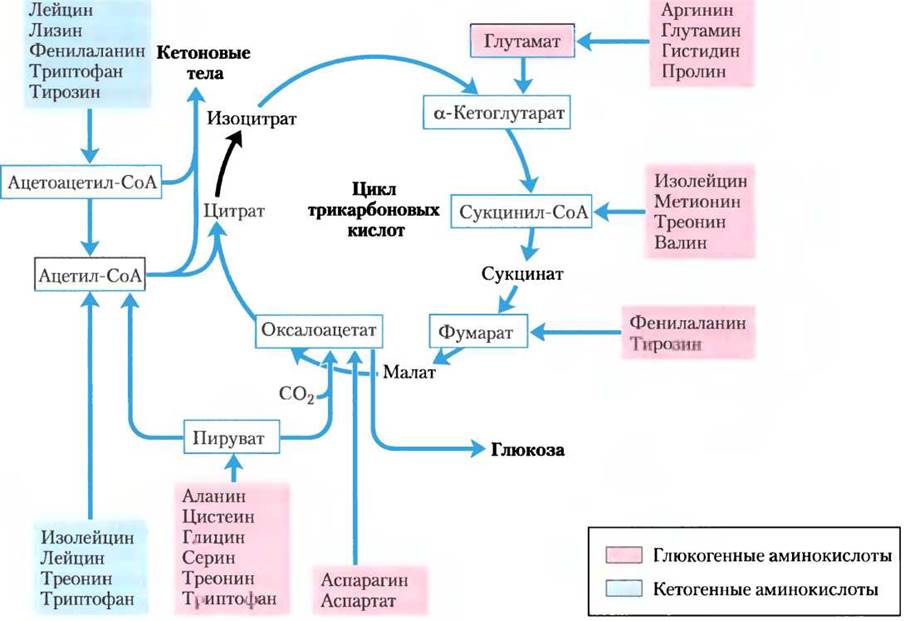
Весь или часть углеродного скелета семи аминокислот в конечном счете разрушаются до ацетил-СоА. Пять аминокислот превращаются в α-кетоглутарат, четыре — в сукцинил-СоА, две — в фумарат и две — в оксалоацетат. Шесть аминокислот целиком или по частям превращаются в пируват, который преобразуется либо в ацетил- СоА, либо в оксалоацетат. Позднее мы суммируем индивидуальные пути метаболизма 20 аминокислот в виде блок-схем, каждый путь приводит к определенному месту входа в цикл трикарбоновых кислот. На этих диаграммах атомы углерода, которые попадают в цикл трикарбоновых кислот, выделены цветом. Обратите внимание, что некоторые аминокислоты встречаются более одного раза, что отражает разные судьбы участков углеродного скелета. Вместо того, чтобы рассматривать отдельные стадии каждого пути аминокислотного катаболизма, мы выбрали для подробного обсуждения некоторые ферментативные реакции, которые особенно примечательны либо из-за их механизма, либо из-за их медицинского значения.
Одни аминокислоты превращаются в глюкозу, другие — в кетоновые тела
Семь аминокислот, которые разлагаются целиком или по частям до ацетоацетил-СоА и/или ацетил- СоА, — это фенилаланин, тирозин, изолейцин, лейцин, триптофан, треонин и лизин. Они могут превратиться в кетоновые тела в печени, где ацетоацетил-СоА превращается в ацетоацетат и затем в ацетон и β-гидроксибутират (см. рис. 17-18). Это кетогенные аминокислоты (рис. 18-15). Их способность образовывать кетоновые тела часто служит маркером для определения неконтролируемого диабета, при котором в печени образуется огромное количество кетоновых тел и из жирных кислот, и из кетогенных аминокислот.
Аминокислоты, которые распадаются с образованием пирувата, α-кетоглутарата, сукцинил- СоА, фумарата и/или оксалоацетата, могут превращаться в глюкозу и гликоген в последовательности реакций, описанных в гл. 14 и 15. Они являются глюкогенными аминокислотами. Разделение на кетогенные и глюкогенные аминокислоты не строгое; пять аминокислот триптофан, фенилаланин, тирозин, треонин и изолейцин выполняют и кетогенные, и глюкогенные функции. Катаболизм аминокислот критичен для выживания животных, потребляющих высокобелковую пищу, и для голодающих животных. Лейцин — исключительно кетогенная аминокислота, широко распространенная в белках. В условиях голодания при его деградации усиливается кетоз.
В катаболизме аминокислот важную роль играют несколько коферментов
В катаболических путях аминокислот встречается широкое разнообразие реакций группировки. Полезно начать изучение этих путей с разбора однотипных реакций, которые не могут протекать без участия кофакторов. Мы уже рассмотрели один важный класс: для реакций трансаминирования необходим пиридоксальфосфат. Другой общий тип реакций в катаболизме аминокислот — это перенос одноуглеродных фрагментов, где обычно участвует один из трех кофакторов: биотин, тетрагидрофолат или S-аденозилметионин (рис. 18-16). Эти кофакторы переносят одноуглеродную группу в различном окисленном состоянии: биотин переносит углерод в его наиболее окисленной форме — как СO2 (см. рис. 14-18); тетрагидрофолат переносит одноуглеродный фрагмент в промежуточном состоянии окисленности и иногда в виде метальной группы; S-аденозилметионин переносит метальную группу — наиболее восстановленное состояние углерода. Последние два кофактора особенно важны в метаболизме аминокислот и нуклеотидов.
Рис. 18-16. Некоторые кофакторы ферментов, имеющие большое значение в реакциях переноса одноуглеродных фрагментов. Атомы азота тетрагидрофолата, к которым присоединяются одноуглеродные группы, показаны голубым.
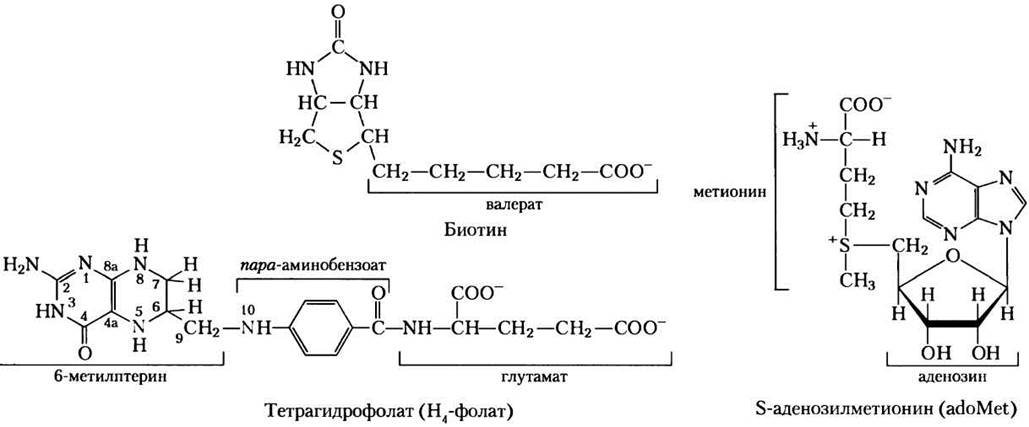
Тетрагидрофолат (Н4-фолат), синтезирующийся бактериями, состоит из замещенного птерина (6-метилптерин), пара-аминобензоата и фрагмента глутамата (рис. 18-16).
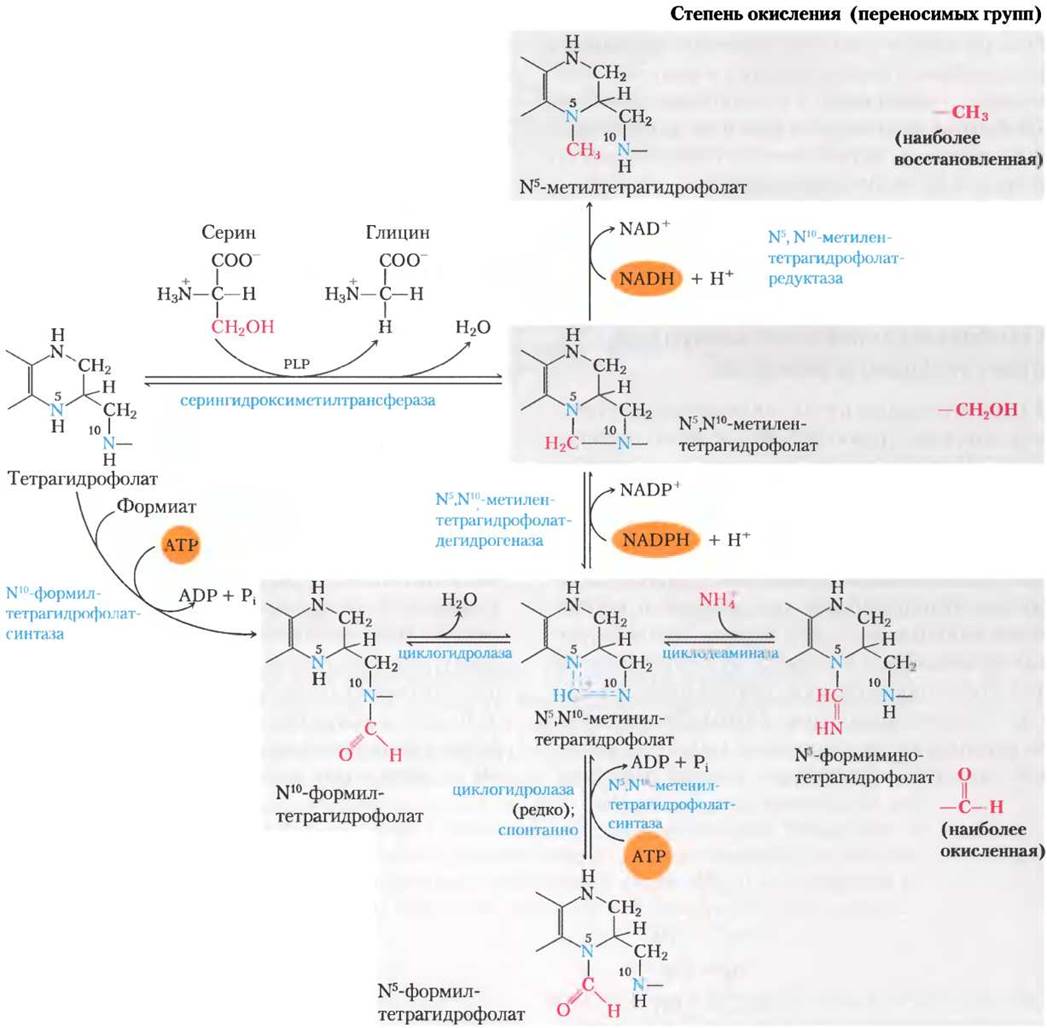
Его окисленная форма, фолат, — витамин для млекопитающих; он может быть в две стадии превращен в тетрагидрофолат под действием фермента дигидрофолатредуктазы. Переносимая одноуглеродная группа в любом из трех окисленных состояний связывается с атомами N-5 или N-10, или с обоими этими атомами азота. Наиболее восстановленная форма кофактора переносит метильную группу, более окисленная — метиленовую группу, а самая окисленная — остаток метинильной группы, формила или формимина (рис. 18-17). Большинство форм тетрагидрофолата подвергаются взаимопревращениям и служат донорами одноуглеродной единицы в разнообразных метаболических реакциях. Главный источник одноуглеродных фрагментов для тетрагидрофолата — это углерод, донором которого является серин. В результате образуются глицин и N5, N10-метилентетрагидрофолат.
Рис. 18-17. Преобразования одноуглеродного фрагмента на тетрагидрофолате. Разные молекулы сгруппированы в соответствии со степенью окисления, наиболее восстановленная форма изображена вверху, а наиболее окисленная — внизу. Все атомы, расположенные на плашке одного цвета, находятся в одинаковом окисленном состоянии. Превращение N5, N10-метилентетрагидрофолата в N5-метилтетрагидрофолат фактически обратимо. При ферментативном переносе формильной группы, как и в пуриновом синтезе (см. рис. 22-33) и образовании формилметионина у бактерий (гл. 27), обычно используется N10-формилтетрагидрофолат вместо N5-формилтетрагидрофолата. Последний значительно более стабилен и поэтому более слабый донор формиминогруппы. N5-Формилтетрагидрофолат — минорный побочный продукт циклогидролазной реакции, а также может образовываться спонтанно. Превращение N5-формилтетрагидрофолата в N5, N10-метенилтетрагидрофолаттребует затраты энергии АТР, потому что иначе равновесие смещено в другую сторону. Обратите внимание на то, что N5-формиминотетрагидрофолат образуется из гистидина в пути, показанном на рис. 18-26.
Хотя тетрагидрофолат может переносить метильную группу на атоме N-5, для осуществления большинства реакций биосинтеза энергии этой метильной группы недостаточно. В качестве переносчика метильной группы лучше подходит кофактор S-аденозилметионин (adoMet). Он синтезируется из АТР и метионина под действием метионинаденозилтрансферазы (рис. 18-18, стадия (1)). Эта реакция необычна тем, что нуклеофильный атом серы метионина вместо атомов фосфора атакует 5'-углерод рибозы молекулы АТР. Трифосфат высвобождается и «разрезается» на Pi и PPi этим же ферментом, a PPi расщепляется неорганической пирофосфатазой; таким образом, в этой реакции разрушаются три связи, включая две связи высокоэнергетических фосфатных групп. Единственная другая известная реакция, в которой трифосфат отщепляется от АТР, протекает при синтезе кофермента В12 (см. доп. 17-2, рис. 3).
Рис. 18-18. Синтез метионина и S-аденозилметионина в метил-активированном цикле. Стадии описаны в тексте. В реакции, катализируемой метионинсинтазой (стадия (4)), метильная группа переносится на кобаламин с образованием метилкобаламина, который в свою очередь выступает донором метила в реакции образования метионина. S-Аденозилметионин, который несет положительно заряженный атом серы (ион сульфония), является мощным метилирующим агентом в ряде реакций биосинтеза. Акцептор метила (стадия (2)) обозначен буквой R.
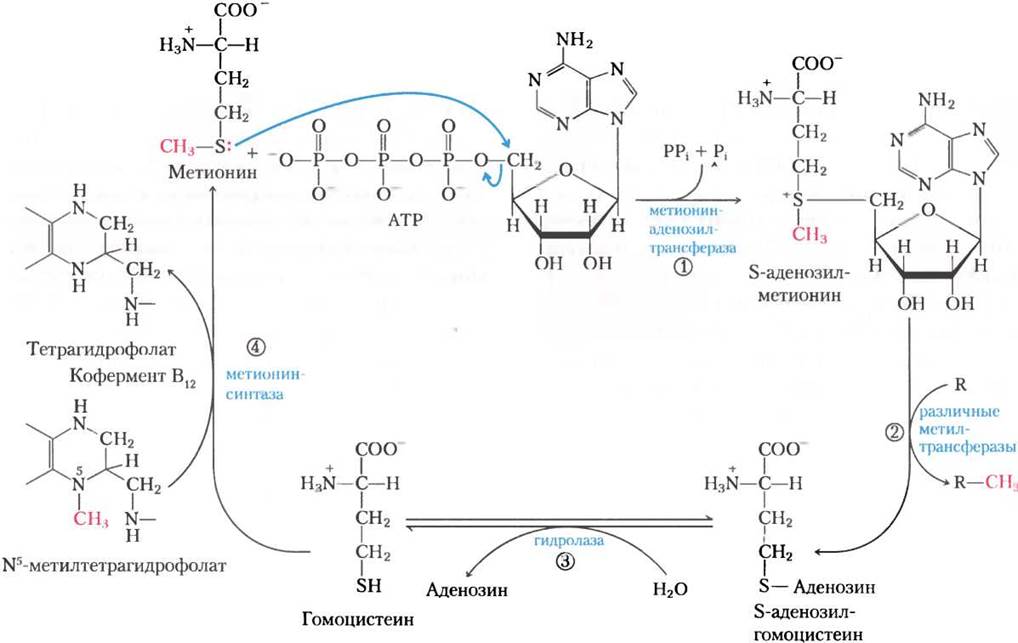
S-Аденозилметионин — мощный алкилирующий агент благодаря дестабилизирующим свойствам его сульфониевого иона. Метильная группа S-аденозилметионина служит мишенью для нуклеофильной атаки — она в 1000 раз более реакционноспособна, чем метильная группа N5-метилтетрагидрофолата.
В результате переноса метильной группы с S-аденозилметионина на акцептор образуется S-аденозилгомоцистеин (рис. 18-18, стадия (2)), который затем расщепляется на гомоцистеин и аденозин (стадия (3)). Метионин регенерируется при переносе метильной группы на гомоцистеин в реакции, катализируемой метионинсинтазой (стадия (4)); так метионин превращается в S-аденозилметионин, что завершает цикл активации метила.
Одна из форм метионинсинтазы, общая для бактерий, использует в качестве донора метила N5-метилтетрагидрофолат. Другая форма фермента, присутствующая у некоторых бактерий и млекопитающих, использует N5-метилтетрагидрофолат, но метильная группа сначала переносится на производное кофермента В12 — кобаламин. Образуется метилколабамин, который служит донором метила при образовании метионина. Эта реакция и перегруппировка L-метилмалонил-СоА до сукцинил-СоА (см. доп. 17-2, рис. 1, а) — единственные известные у млекопитающих реакции, зависимые от кофермента В12.
В этих метаболических путях витамин В12 и фолат тесно связаны между собой. Недостаточность витамина В12 (пернициозная анемия) — редкое заболевание, которое встречается лишь у людей с нарушением всасывания этого витамина в кишечнике (см. доп. 17-2) или у строгих вегетарианцев (витамина В12 нет в растительных клетках). Болезнь развивается медленно, поскольку организму требуются совсем небольшие количества витамина В12, а его запасов в печени хватает на период от трех до пяти лет. Симптомами заболевания являются не только анемия, но и различные неврологические нарушения.
Анемия может быть связана с реакцией, катализируемой метионинсинтазой. Как упоминалось выше, метильная группа метилкобаламина происходит из N5- метилтетрагидрофолата, и в клетках млекопитающих это единственная реакция, в которой используется N5-метил- тетрагидрофолат. Превращение N5, N10-метиленовой формы в N5-метильную форму тетрагидрофолата происходит необратимо (рис. 18-17). Поэтому, если кофермент В12 недоступен для синтеза метилкобаламина, происходит захват фолата в N5-метильной форме. Анемия, связанная с неусвоением витамина В12, называется мегалобластной анемией. Она сопровождается
снижением содержания зрелых эритроцитов и появлением в костном мозге незрелых клеток- предшественников, или мегалобластов. В крови эритроциты постепенно замещаются меньшим количеством аномально крупных эритроцитов, называемых макроцитами. Дефект развития эритроцитов является прямым следствием исчерпания запаса N5, N10-метилентетрагидрофолата, необходимого для синтеза тимидиновых нуклеотидов, входящих в состав ДНК (см. гл. 22). Дефицит фолата, при котором исчерпаны запасы всех форм тетрагидрофолата, также приводит к анемии, причины которой приблизительно те же. Симптомы недостаточности витамина В12 можно сгладить, принимая либо сам витамин, либо фолат.
Однако лечить пернициозную анемию одними только препаратами фолиевой кислоты опасно, поскольку неврологические симптомы дефицита витамина В12 будут прогрессировать. Эти симптомы не связаны с дефектом метионин- синтетазы. В данном случае аномальный фермент метилмалонил-СоА-мутаза вызывает накопление в мембранах нейронов необычной жирной кислоты, в составе которой нечетное число атомов углерода. Поэтому при анемии, связанной с недостаточностью фолата, часто назначают как фолат, так и витамин В12, по крайней мере, в тех случаях, когда точная метаболическая причина болезни не установлена. Ранняя диагностика дефицита витамина В12 имеет очень большое значение, поскольку некоторые связанные с этим неврологические нарушения могут быть необратимыми.
Недостаток фолата также сопровождается уменьшением количества N5-метилтетра- гидрофолата, необходимого для реакции метионинсинтетазы. Это приводит к повышению уровня гомосерина в крови, что может спровоцировать развитие инфаркта миокарда, гипертензию и инсульт. По оценкам, высокий уровень гомосерина является причиной 10% случаев инфаркта миокарда. Данное нарушение корректируется приемом фолиевой кислоты. ■
Другой кофактор катаболизма аминокислот тетрагидробиоптерин похож на фрагмент птерина тетрагидрофолата, но он не вовлечен в реакции переноса одноуглеродных фрагментов; вместо этого он участвует в оксилительных реакциях. Мы рассмотрим его действие, когда будем обсуждать деградацию фенилаланина (см. рис. 18-24).
Шесть аминокислот расщепляются до пирувата
Углеродные скелеты шести аминокислот целиком или частично превращаются в пируват. Пируват может образовывать либо ацетил-СоА и рано или поздно окисляться в цикле трикарбоновых кислот, либо оксалоацетат и поступать в глюконеогенез. Вот эти шесть аминокислот — аланин, триптофан, цистеин, серин, глицин и треонин (рис. 18-19). Аланин просто превращается в пируват в реакции трансаминирования с α-кетоглутаратом, а боковая цепь триптофана отщепляется в виде аланина, а значит, в итоге пирувата.Цистеин перегруппировывается в пируват в две стадии: сначала удаляется атом серы, а затем происходит трансаминирование. Серин превращается в пируват под действием сериндегидратазы. В этой пиридоксальфосфат- зависимой реакции удаляются и β-гидроксил, и α-аминогруппа серина (рис. 18-20, а).
Глицин расщепляется тремя путями и только один из них ведет к пирувату. Глицин превращается в серин путем ферментативной реакции присоединения гидроксиметильной группы (рис. 18-19 и 18-20, б). Для этой реакции, катализируемой серин-гидроксиметил-трансферазой, необходимы коферменты тетрагидрофолат и пиридоксальфосфат. Серин переводится в пируват, как было описано выше. По второму пути, который у животных встречается чаще, глицин подвергается окислительному расщеплению до СО2, NH+4 и метиленовой группы (-СН2-) (рис. 18-19, 18-20, в). Для этой легко обратимой реакции, катализируемой глицинрасщепляющим ферментом (глицинсинтазой), также необходим тетрагидрофолат, который принимает метиленовую группу. В этом пути окислительного расщепления два атома углерода не попадают в цикл трикарбоновых кислот. Один атом теряется в виде СO2, а второй превращается в метиленовую группу N5, N10- метилентетрагидрофолата (рис. 18-17), который в определенных путях биосинтеза выступает как донор одноуглеродного фрагмента. Работа этого второго пути расщепления глицина, вероятно, критична для млекопитающих. Люди с серьезными дефектами в активности глицин-расщепляющего фермента страдают заболеванием, известным как некетотическая гиперглицинемия. Эта болезнь характеризуется повышенным уровнем глицина в сыворотке, что приводит к серьезным нарушениям умственного развития и смерти в раннем детстве. Высокие концентрации глицина — тормозного нейромедиатора — одно из возможных объяснений неврологических последствий этой болезни. У людей было определено множество разных генетических дефектов в метаболизме аминокислот (табл. 18-2). В этой главе мы встретимся еще с некоторыми из них. ■
Рис. 18-19. Катаболические пути аланина, глицина, серина, цистеина, триптофана и треонина. Судьба индольной группы триптофана показана на рис. 18-21. Детали большинства реакций, в которых участвуют серин и глицин, показаны на рис. 18-20. Изображенный здесь путь расщепления треонина включает только третью часть полного катаболизма треонина (альтернативные пути см. на рис. 18-27). Некоторые пути деградации цистеина ведут к пирувату. Сера, входящая в состав цистеина, может участвовать в разных путях, один из которых изображен на рис. 22-15. Атомы углерода здесь и в следующих рисунках окрашены в разные цвета, чтобы проследить их судьбу в молекулах.
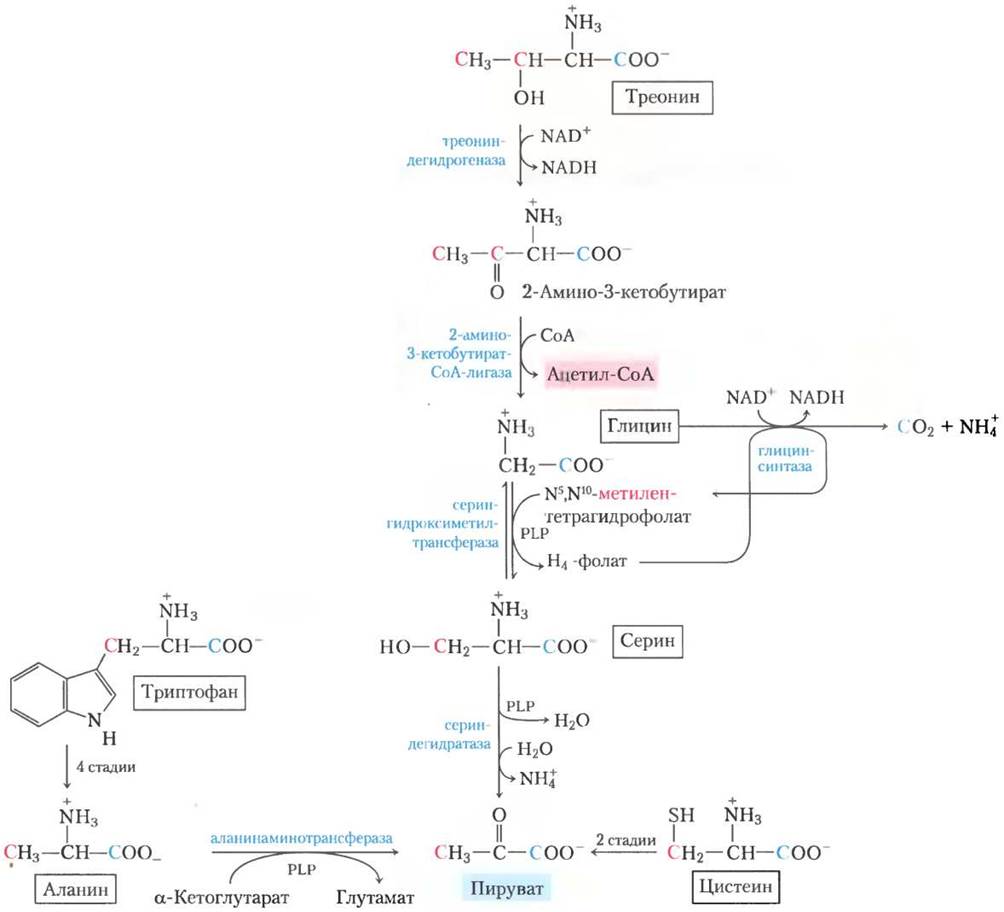
Рис. 18-20. Механизм реакции. Роль кофакторов пиридоксальфосфата и тетрагидрофолата в метаболизме глицина и серина. Первая стадия каждой реакции (не показана) представляет собой образование ковалентной иминной связи между PLP, связанным с ферментом, и субстратом — аминокислотой серином в случае (а), глицином в (б) и (в), а — катализируемое PLP отщепление воды в сериндегидрогеназной реакции (стадия (1)) ведет к последующему образованию пирувата. б — в гидрокси- метилтрансферазной реакции стабилизированный PLP- карбанион (продукт стадии (1)) является ключевым интермедиатом в переносе метиленовой группы (в как -СН2-OН) с N5, N10-метилентетрагидрофолата с образованием серина. в — глицинрасщепляющий фермент (глицинсинтаза) представляет собой полиферментный комплекс из субъединиц P, H, Т и L. В конечном итоге в этой обратимой реакции выделяются СO2 и NH+4, а второй атом углерода глицина присоединяется к тетрагидрофолату с образованием N5, N10-метилентетрагидрофолата. Пиридоксальфосфат активирует α-углерод аминокислот на критических стадиях всех этих реакций, а тетрагидрофолат переносит одноуглеродные фрагменты в двух из них (см. рис. 18-6,18-17).

Таблица 18-2. Некоторые генетические болезни, вызванные дефектами в катаболизме аминокислот
Заболевание |
Примерная частота встречаемости (на 100 000 новорожденных) |
Дефектный процесс |
Дефектный фермент |
Симптомы и эффекты |
Альбинизм |
<3 |
Синтез меланина из тирозина |
Тирозин 3-монооксигеназа (тирозиназа) |
Отсутствие пигментации: белые волосы, белая кожа |
Алкаптонурия |
<0,4 |
Деградация тирозина |
Гомогентизат- 1,2-диоксигеназа |
Черный цвет мочи; развивающийся позднее артрит |
Аргининемия |
<0,5 |
Синтез мочевины |
Аргиназа |
Задержка умственного развития |
Аргининосукцинатная ацидемия |
<1,5 |
Синтез мочевины |
Аргининосукциназа |
Рвота; конвульсии |
Дефицит карбамоил- фосфатсинтетазы I |
<0,5 |
Синтез мочевины |
Карбамоилфосфат- синтетаза I |
Летаргия; конвульсии; ранняя смерть |
Гомоцистинурия |
<0,5 |
Деградация метионина |
Цистатионин- β-синтаза |
Дефектное развитие костей: задержка умственного развития |
Болезнь кленового сиропа (кетоацидурия) |
<0,4 |
Распад изолейцина, лейцина и валина |
Дегидрогеназный комплекс Разветвленных α-кетокислот |
Рвота; конвульсии; задержка умственного развития; ранняя смерть |
Метилмалоновая ацидемия |
<0,5 |
Превращение пропионил-СоА в сукцинил-СоА |
Метилмалонил- СоА- мутаза |
Рвота; конвульсии; задержка умственного развития; ранняя смерть |
Фенил кетонурия |
<8 |
Превращение фенилаланина в тирозин |
Фенилаланин-гидроксилаза |
Рвота у новорожденных; задержка умственного развития |
В третьем, и последнем, пути деградации глицина ахиральная молекула глицина служит субстратом для фермента оксидазы D-аминокислот. Глицин превращается в глиоксилат — альтернативный субстрат для печеночной лактатдегидрогеназы (с. 91). Глиоксилат окисляется в NAD+- зависимой реакции до оксалоацетата:
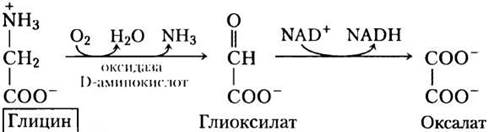
Основная функция оксидазы D-аминокислот, присутствующей в больших количествах в почках, — это детоксикация поглощенных производных D-аминокислот, образующихся из клеточной стенки бактерий и продуктов питания (высокая температура при жарке вызывает спонтанную рацемизацию небольшого количества L-аминокислот в белках). Оксалат, поступающий с пищей или ферментативно образующийся в почках, влияет на здоровье человека. Кристаллы оксалата кальция составляют до 75% всех камней в почках. ■
Существуют два главных пути распада треонина. Один путь ведет к образованию пирувата через глицин (рис. 18-19). Превращение до глицина происходит в две стадии, при этом сначала под действием треониндегидрогеназы треонин превращается в 2-амино-З-кетобутират. На этот путь приходится всего лишь от 10% до 30% всего катаболизма треонина у человека, но для других млекопитающих он может иметь большее значение. У человека главный путь ведет к образованию сукцинил-СоА, и он будет описан ниже.
В лабораторных условиях серин-гидрокси- метилтрансфераза может катализировать превращение треонина до глицина и ацетальдегида в одну стадию, но у млекопитающих этот путь практически не активен.
Семь аминокислот распадаются до ацетил-СоА
Части углеродных скелетов семи аминокислот триптофана, лизина, фенилаланина, тирозина, лейцина, изолейцина и треонина образуют ацетил-СоА и/или ацетоацетил-СоА, последний переводится в ацетил-СоА (рис. 18-21). Некоторые последние стадии деградации лейцина, лизина и триптофана напоминают стадии окисления жирных кислот. Треонин (не показан на рис. 18-21) превращается в ацетил- СоА во второстепенном пути, изображенном на рис. 18-19.
Рис. 18-21. Катаболические пути триптофана, лизина, фенилаланина, тирозина, лейцина и изолейцина. Некоторые атомы углерода этих аминокислот (выделены красным) превращаются в ацетил-СоА. Триптофан, фенилаланин и изолейцин также содержат атомы углерода (показаны синим), которые становятся частью пирувата или другими интермедиатами цикла трикарбоновых кислот. Путь деградации фенилаланина более детально изображен на рис. 18-23. Судьба атомов азота на этой схеме не показана, в большинстве случаев они переносятся на α-кетоглутарат с образованием глутамата.
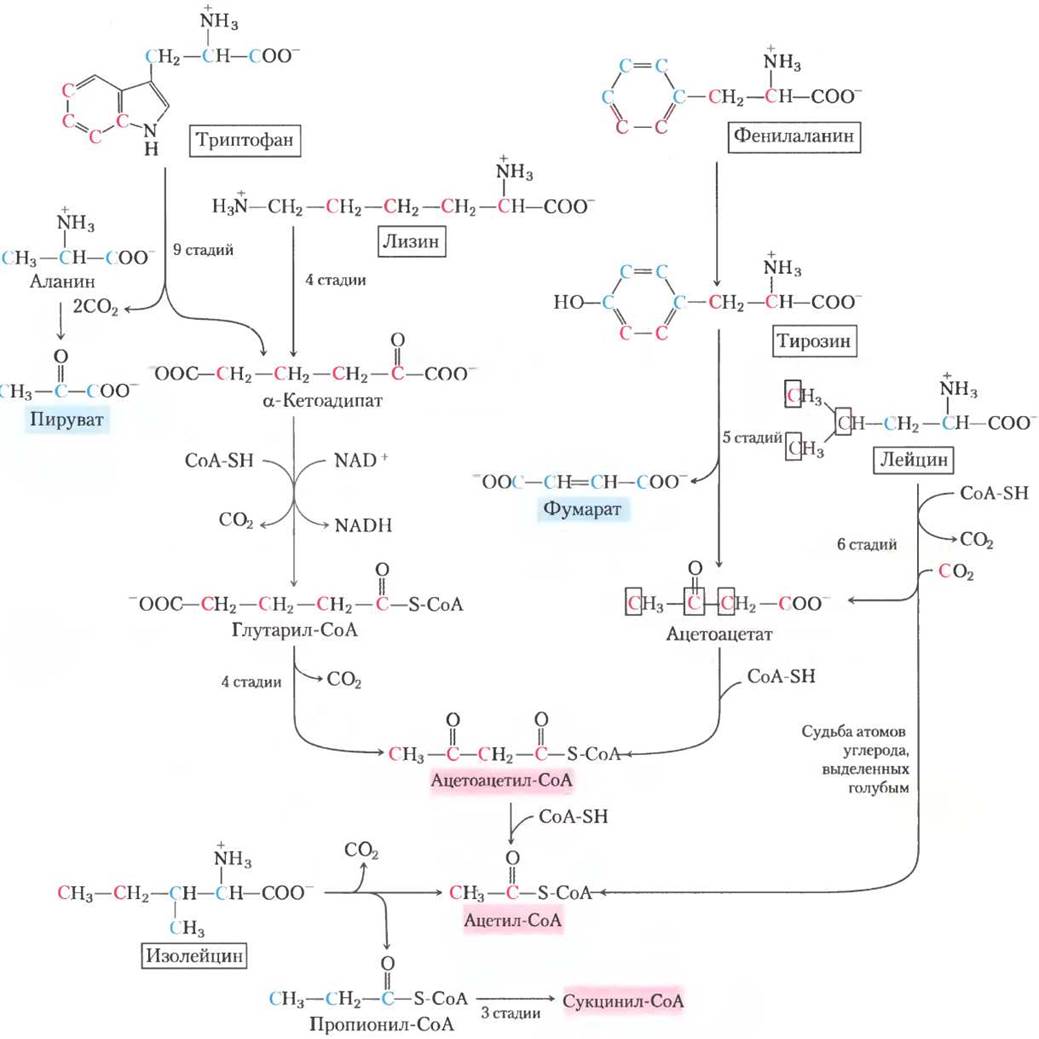
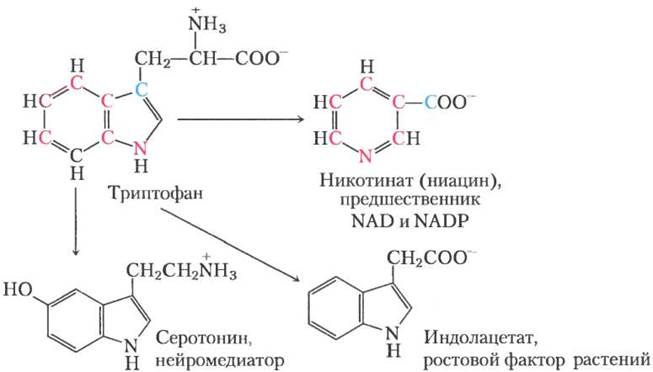
Катаболитические пути двух из этих семи аминокислот заслуживают особого внимания. Триптофан расщепляется в наиболее сложном из всех путей аминокислотного катаболизма в тканях животных; часть триптофана (четыре из его атомов углерода) превращается в ацетил-СоА через ацетоацетил-СоА. Некоторые интермедиаты катаболизма триптофана являются предшественниками в синтезе других биомолекул (рис. 18-22), включая никотинат — предшественник NAD и NADP у животных; серотонин — нейромедиатор позвоночных; и индолацетат — ростовой фактор растений. Некоторые из этих путей биосинтеза описаны более детально в гл. 22 (см. рис. 22-28,22-29).
Рис. 18-22. Триптофан — предшественник некоторых биологически важных веществ. Из ароматического кольца триптофана синтезируются никотинат (ниацин), индолацетат и серотонин. Атомы, из которых получается кольцо никотината, выделены цветом.
Генетические дефекты в ферментах, участвующих в путях распада фенилаланина, ведут к некоторым наследственным заболеваниям человека (рис. 18-23); это мы обсудим далее. Фенилаланин и продукт его окисления тирозин (оба содержат по девять атомов углерода) распадаются на два фрагмента, которые могут войти в цикл трикарбоновых кислот: четыре из девяти атомов углерода в составе ацетоацетата, который превращается в ацетоацетил-СоА и затем в ацетил-СоА, а второй четырехуглеродный фрагмент — в фумарат. Итак, восемь из девяти атомов углерода этих двух аминокислот вовлекаются в цикл трикарбоновых кислот, девятый теряется в виде СО2. После реакции гидроксилирования фенилаланин превращается в тирозин, который является также предшественником нейромедиатора дофамина и гормонов норадреналина и адреналина, секретируемых мозговым веществом надпочечников (см. рис. 22-29). Кроме того, из тирозина образуется меланин, черный пигмент кожи и волос.
Рис. 18-23. Катаболические пути фенилаланина и тирозина. В норме у человека эти аминокислоты превращаются в ацетоацетил-СоА и фумарат. Генетические дефекты многих ферментов (показаны желтым цветом) вызывают врожденные заболевания.

Катаболизм фенилаланина у некоторых людей может быть нарушен
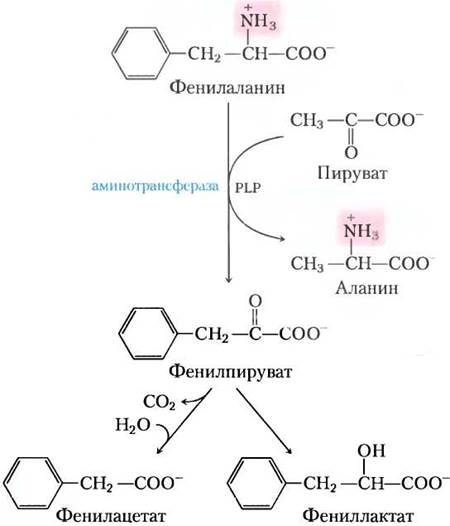
Многие аминокислоты являются нейромедиаторами, предшественниками медиаторов или антагонистами нейромедиаторов, поэтому генетические дефекты метаболизма аминокислот могут вызывать нервные расстройства и задержку умственного развития. При большинстве таких заболеваний накапливается специфический интермедиат. Например, дефекты в гене фенилаланингидролазы, первого фермента в катаболизме фенилаланина (рис. 18-23), вызывают болезнь фенилкетонурию, наиболее распространенную причину повышенного уровня фенилаланина (гиперфенилаланинемия).
Фенилаланингидроксилаза (фенилаланин- 4-монооксигеназа) принадлежит к одному из основных классов ферментов, которые называются оксидазами со смешанными функциями (см. доп. 21-1). Они катализируют спонтанное гидроксилирование субстрата одним атомом кислорода молекулы О2 с восстановлением другого атома до Н2О. Фенилаланингидроксилаза использует в качестве кофактора тетрагидробиоптерин, который переносит электроны с NАDН на O2 и окисляется в этом процессе до дигидробиоптерина (рис. 18-24). Впоследствии он восстанавливается ферментом дигидробиоптеринредуктазой в реакции, для которой необходим NАDН.
Рис. 18-24. Роль тетрагидробиоптерина в реакции, катализируемой фенилаланингидроксилазой. Атом водорода, выделенный розовым, переносится прямо с С-4 на С-3. Эта особенность, открытая в Национальных институтах здравоохранения США (National Institutes of Health, сокращенно NIH), была названа NIH-сдвигом.
При фенилкетонурии активируется вторичный малоиспользуемый в норме путь метаболизма фенилаланина. В этом пути фенилаланин подвергается трансаминированию с пируватом с образованием фенилпирувата (рис. 18-25).
Рис. 18-25. Альтернативные пути катаболизма фенилаланина при фенилкетонурии. При фенилкетонурии в тканях, крови и моче накапливается фенилпируват. Моча может также содержать фенилацетат и фениллактат.
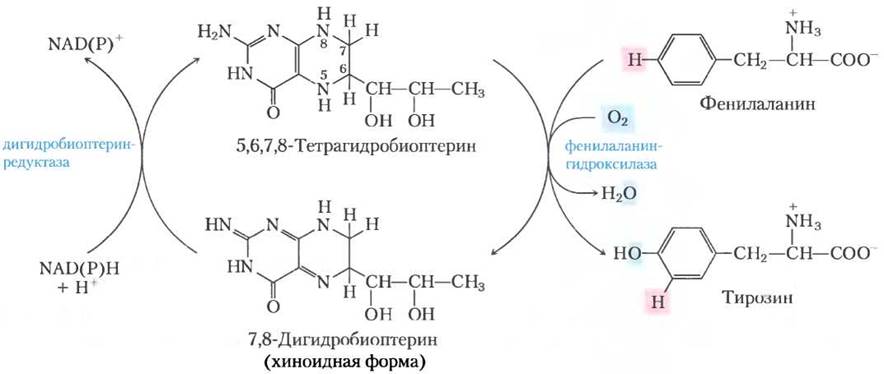
Фенилаланин и фенилпируват накапливаются в крови и тканях и выделяются с мочой — отсюда и название «фенилкетонурия». Большая часть фенилпирувата вместо того, чтобы выводиться, либо декарбоксилируется до фенилаце- тата, либо восстанавливается до фениллактата. Фенилацетат придает характерный запах моче, по которому обычно и определяют фенилкетонурию у новорожденных. Накопление фенилаланина или его метаболитов в раннем возрасте ухудшает нормальное развитие мозга, вызывая серьезную задержку в умственном развитии. Это может быть вызвано избытком фенилаланина, который конкурирует с другими аминокислотами за переносчик этой аминокислоты из крови в клетки мозга, что в итоге приводит к дефициту необходимых метаболитов.
Фенилкетонурия — одно из первых наследственных нарушений метаболизма, обнаруженных у людей. Если эту проблему распознать в раннем детстве, развитие умственной отсталости можно предотвратить строго контролируемой диетой. В пище должно содержаться только необходимое для синтеза белков количество фенилаланина и тирозина. Потребление пищи, богатой белками, надо ограничить. Такие природные белки, как казеин молока, сначала должны быть гидролизированы, а основные количества фенилаланина удалены для того, чтобы обеспечить необходимую диету, хотя бы в детстве. Искусственный подсластитель аспартам представляет собой дипептид аспартата и метилового эфира фенилаланина (см. рис. 1-23, б), поэтому пища, подслащенная аспартамом, опасна для человека, вынужденного соблюдать диету по фенилаланину.
Фенилкетонурия может быть вызвана также дефектом фермента, который катализирует регенерацию тетрагидробиоптерина (рис. 18-24). Лечение в этом случае более сложное, чем ограничение потребления фенилаланина и тирозина. Тетрагидробиоптерин требуется и для образования L-3,4-дигидроксифенилаланина (L-дофа) и 5-гидрокситриптофана — предшественников нейромедиаторов норадреналина и серотонина. При этой форме фенилкетонурии эти предшественники необходимо включать в диету. Добавление самого тетрагидробиоптерина в пищу неэффективно, потому что он нестабилен и не проходит гематоэнцефалический барьер, а значит, не включается в фермент.
Диагностика генетических болезней у новорожденных может быть довольно эффективной, особенно в случае фенилкетонурии. Проводимые тесты (не просто запах мочи!) не слишком дорогие, а определение и раннее лечение фенилкетонурии в младенческом возрасте (выявляется от восьми до десяти случаев на 100 000 новорожденных) ежегодно сохраняют миллионные суммы, которые требуются при запаздалом лечение заболевания. И, что более важно, ранее обнаружение заболевания с помощью этих несложных тестов дает возможность избежать эмоциональной травмы, а это уже трудно оценить деньгами.
Другое наследственное нарушение катаболизма фенилаланина — алкаптонурия, при которой дефектен фермент гомогентизатдиоксигеназа (рис. 18-23). Это заболевание считается менее серьезным, чем фенилкетонурия, оно вызывает менее трагические последствия, но надо знать, что из-за выделения большого количества гомогентизата, который окисляется, моча больного становится черной. У больных алкаптонурией также вероятно развитие некоторых форм артрита. Алкаптонурия представляет исторический интерес. В начале 1900-х гг. Арчибальд Гаррод открыл, что это заболевание наследственное. Он выяснил, что причиной является отсутствие одного фермента. Гаррод был первым, кто выявил связи между наследственным заболеванием и ферментом, что было большим прорывом на пути развития науки и неизбежно вело к современному пониманию генов и информационных путей, описанных в части III. ■
Пять аминокислот превращаются в α-кетоглутарат
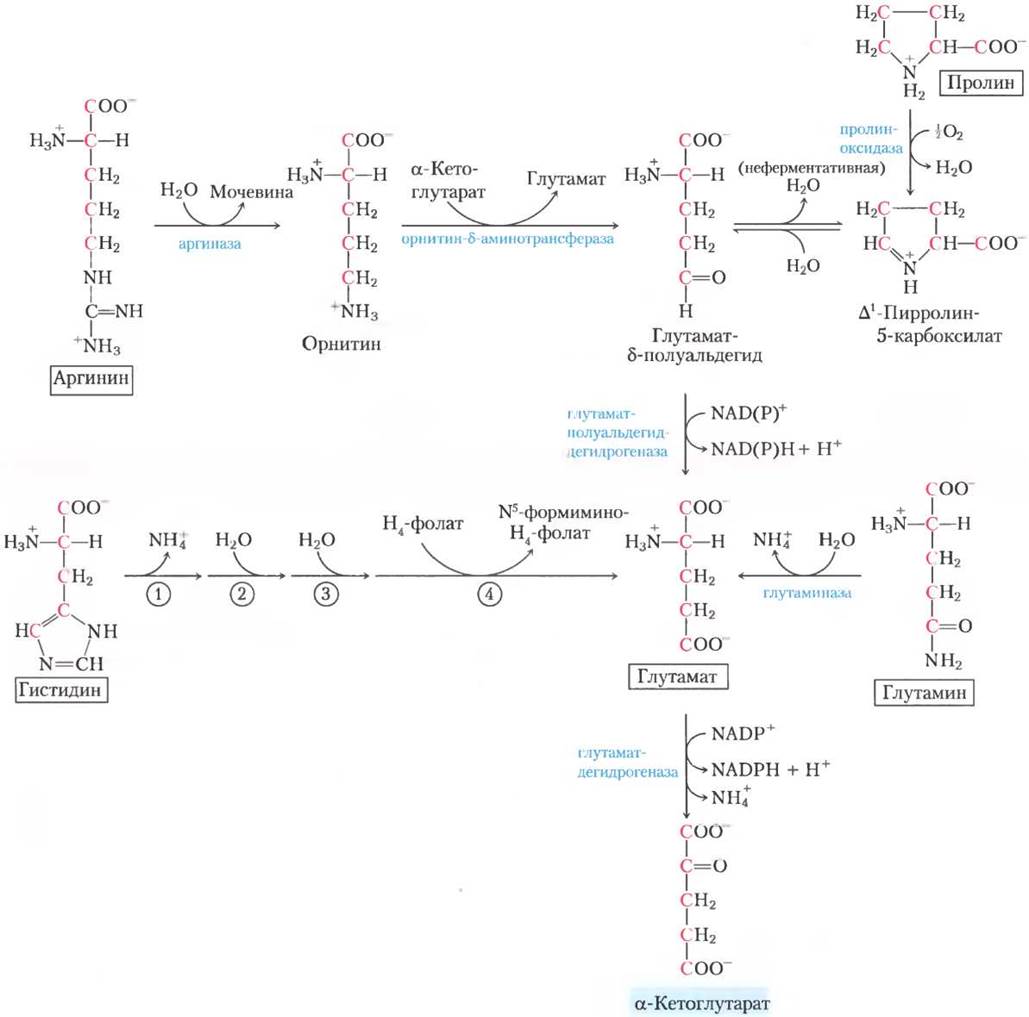
Углеродный скелет пяти аминокислот (пролин, глутамат, глутамин, аргинин и гистидин) попадают в цикл трикарбоновых кислот в виде α-кетоглутората (рис. 18-26). Пролин, глутамат и глутамин содержат по пять атомов углерода. Циклическая структура пролина раскрывается при окислении наиболее удаленного от карбоксильной группы углерода с образованием шиффова основания, после гидролиза которого из исходного пролина получается линейный глутамат-y- полуальдегид. Этот интермедиат далее окисляется на том же атоме углерода с образованием глутамата. При действии глутаминазы или в любой из нескольких ферментативных реакций, в которых глутамин отдает амидный азот на акцептор, глутамин превращается в глутамат. Трансаминирование или дезаминирование глутамата дает α-кетоглутарат.
Аргинин и гистидин содержат цепочку из пяти углеродов, к которой шестой углерод присоединен через атом азота. Поэтому катаболические превращения этих аминокислот в глутамат более сложные, чем путь от пролина до глутамина (рис. 18-26). Аргинин превращается в пятиуглеродный скелет орнитина в цикле мочевины (рис. 18-10), и далее орнитин трансаминируется до глутамат-y-полуальдегида. Превращение гистидина в пятиуглеродный глутамат многостадийное; лишний углерод удаляется в реакции, где в качестве кофактора используется тетрагидрофолат.
Рис. 18-26. Катаболические пути аргинина, гистидина, глутамата, глутамина и пролина. Эти аминокислоты превращаются в α-кетоглутарат. Пронумерованные стадии гистидиного пути катализируются (1) гистидин-аммиак-лиазой, (2) уроканазой, (3) имидазолонпропионазой и (4) глутамат-формиминотрансферазой.
Превращение четырех аминокислот происходит до сукцинил-СоА
Углеродные скелеты метионина, изолейцина, треонина и валина расщепляются по пути, который приводит к сукцинил-СоА (рис. 18-27) — интермедиату цикла трикарбоновых кислот. Метионин отдает свою метильную группу одному из нескольких возможных акцепторов через S-аденозилметионин, а три из четырех оставшихся атомов углерода попадают в пропионат пропионил-СоА — предшественник сукцинил- СоА. Изолейцин подвергается трансаминированию и последующему окислительному декарбоксилированию образующейся α-кетокислоты.
Рис. 18-27. Катаболические пути метионина, изолейцина, треонина и валина. Превращение этих аминокислот идет до сукцинил-СоА; кроме того, из двух атомов углерода изолейцина образуется ацетил-СоА (см. рис. 18-21). Путь деградации треонина, показанный здесь, присутствует у людей; путь, обнаруженный у других организмов, показан на рис. 18-19. Цепочка превращений метионина в гомоцистеин более детально описана на рис. 18-18; гомоцистеина в α-кетобутират — на рис. 22-14; а пропионил-СоА в сукцинил-СоА — на рис. 17-11.
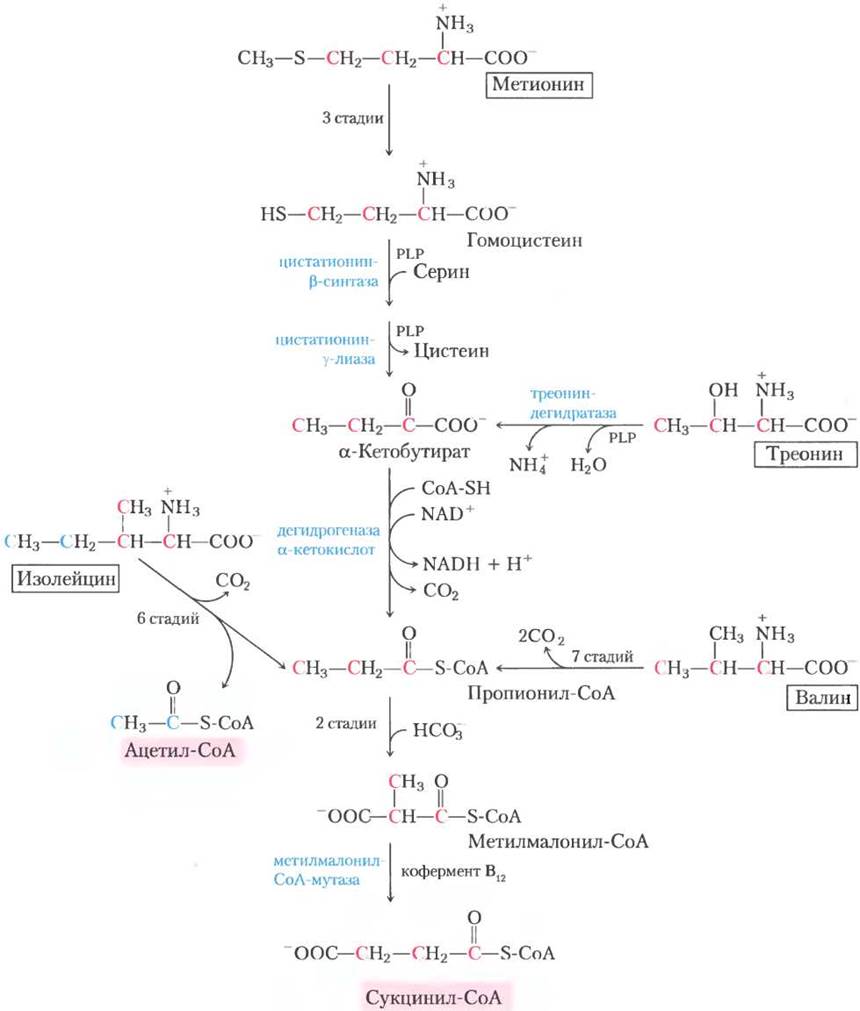
Оставшийся пятиуглеродный скелет в дальнейшем окисляется до ацетил-СоА и пропионил- СоА. Валин претерпевает трансаминирование и декарбоксилирование, затем серию окислительных реакций, которые переводят оставшиеся четыре атома углерода в пропионил-СоА. Некоторые участки путей распада валина и изолейцина похожи на стадии деградации жирных кислот (см. рис. 17-8, а). В тканях человека треонин также в две стадии преобразуется в пропионил- СоА. Это главный путь деградации треонина у людей (альтернативный путь см. на рис. 18-19). Механизм первой реакции аналогичен механизму реакции, катализируемой сериндегидратазой; в действительности же может оказаться, что сериндегидратаза и треониндегидратаза — один фермент.
Пропионил-СоА, образующийся из этих трех аминокислот, превращается в сукцинил-СоА по пути, описанному в гл. 17: карбоксилирование до метилмалонил-СоА, эпимеризация метилмалонил-СоА и превращение в сукцинил-СоА зависимым от В12 ферментом метилмалонил-СоА- мутазой (см. рис. 17-11). При редкой генетической болезни, известной как метилмалоновая ацидемия, метилмалонил-СоА-мутаза отсутствует, что имеет серьезные последствия для обмена веществ (табл. 18-2, доп. 18-2).
Дополнение 18-2. МЕДИЦИНА. Ученые нашли объяснение загадочного убийства
Порой правда бывает более странной, чем выдумка, и больше подходит для сюжета кинофильма. Вот к примеру случай Патрисии Сталлингс. Она была приговорена к пожизненному заключению за убийство своего новорожденного сына, но позднее, благодаря медицинской экспертизе трех упорных ученых, была оправдана.
История началась летом 1989 г., когда Сталлингс доставила своего трехмесячного сына Райна в реанимацию детской больницы им. кардинала Гленнона в Сент- Луисе. У ребенка было затруднено дыхание, неконтролируемая рвота и расстройство желудка. По симптомам лечащий врач-токсиколог поставил диагноз «отравление этиленгликолем» (это компонент антифриза). Это заключение в дальнейшем было подтверждено результатом анализа в коммерческой лаборатории.
Когда ребенок пришел в себя, его отдали в дом малютки, а Сталлингс и ее мужу Дэвиду разрешили навещать мальчика. Но ребенок заболел, а затем умер после того, как Сталлингс побыла с ним наедине короткое время, ее обвинили в преднамеренном убийстве и арестовали без возможности внесения залога. Результаты анализов, полученных в коммерческой лаборатории и в лаборатории больницы, невозможно было оспорить: обе лаборатории обнаружили большие количества этиленгликоля в крови мальчика и следовые количества этого вещества в бутылке молока, которым Сталлингс кормила своего сына во время визита.
Но как ни удивительно, сама того не зная, Сталлингс поставила блестящий эксперимент. Находясь в заключении, она забеременела и в феврале 1990 г. родила другого сына Дэвида Сталлингса мл. Его немедленно передали в дом малютки, но через две недели у него начали появляться симптомы, похожие на симптомы у Райна. Дэвиду сразу же поставили диагноз редкого метаболического расстройства — метилмалоновой ацидемии (ММА). Это рецессивное генетическое нарушение метаболизма аминокислот случается примерно у одного из 48 000 новорожденных, причем симптомы этой болезни практически идентичны симптомам, вызванным отравлением этиленгликолем.
Сталлингс не имела возможности отравить своего второго сына, но прокурорам шт. Миссури сложно было принять к рассмотрению новые технологии исследований, поэтому они настаивали на проведении суда над ней в любом случае. Суд не позволил использовать в качестве доказательства диагноз ММА у второго ребенка, и в январе 1991 г. Патрисии Сталлингс было предъявлено обвинение в преднамеренном убийстве, она была приговорена к пожизненному заключению.
К счастью для Сталлингс, Уильям Слай, заведующий кафедрой биохимии и молекулярной биологии Университета Сент-Луиса, и Джеймс Шумейкер, руководитель лаборатории скрининга метаболитов в этом университете, заинтересовались этим случаем, узнав о нем из телепередачи. Шумейкер сам переделал анализ крови Райна и не обнаружил там этиленгликоля. Ученые связались с Пьеро Ринальдо, экспертом по метаболическим болезням в Медицинской школе Йельского университета — он имел лабораторию, оборудованную для диагностики ММА по пробам крови.
Сделав анализ крови Райна, Ринальдо нашел высокие концентрации метилмалоновой кислоты, продукта распада разветвленных аминокислот изолейцина и валина; метилмалоновая кислота накапливается у больных ММА из-за дефекта в ферменте, который должен превращать ее в следующий продукт метаболического пути. И особенно примечательно, как он сказал, что кровь и моча ребенка содержали огромное количество кетонов — других метаболических свидетельств этой болезни. Как и Шумейкер, он не нашел этиленгликоля в образцах жидкостей организма ребенка. Молоко для вскармливания из бутылочки протестировать не удалось, потому что бутылочка исчезла таинственным образом. Анализ, проведенный Ринальдо, убедил его, что Райн умер от ММА. Но как же результаты двух лабораторий, показавших наличие этиленгликоля в его крови? Неужели в обоих случаях была допущена ошибка?
Ринальдо изучил документацию лабораторий и увидел, по его словам, «ужасающее». Первая лаборатория показала, что образец крови Райна Сталлингса содержал этиленгликоль, даже при том, что анализ крови не соответствовал анализу контрольной пробы, содержащей этиленгликоль. «Это не просто вопрос сомнительной интерпретации результатов анализа. Качество их анализа просто неприемлемо», — сказал Ринальдо. А что со второй лабораторией? Как и Ринальдо, эта лаборатория обнаружила аномальные вещества в крови Райна и просто «приняла, что это этиленгликоль». «Хотя в образцах из бутылочки не было обнаружено ничего необычного, — возмущался Ринальдо, — все же лаборатория заявила о присутствии этиленгликоля там тоже».
Ринальдо представил свои результаты обвинителю Джоржу МакЭлрою, который на следующий день на пресс-конференции заявил: «Я больше не верю экспертизам этих лабораторий». Рассмотрев заключение о том, что Райн Сталлингс умер от ММА, 20 сентября 1991 г. МакЭлрой снял все обвинения с Патрисии Сталлингс.
По материалам Мишель Хофман (1991). Science 253,931. (С) American Association for the Advancement of Science, 1991
Разветвленные аминокислоты не деградируют в печени
Катаболизм большинства аминокислот происходит в печени, три аминокислоты с боковыми цепями (лейцин, изолейцин и валин) окисляются для получения энергии главным образом в мышцах, жировой ткани, почках и в мозге. Эти внепеченочные ткани содержат аминотрансферазу, которая отсутствует в печени. Этот фермент превращает все три разветвленных аминокислоты в соответствующую α-кетокислоту (рис. 18-28). Затем дегидрогеназный комплекс разветвленных α-кетокислот катализирует окислительное декарбоксилирование всех трех α-кетокислот, в каждом случае высвобождая карбоксильную группу в виде СО2 и образуя ацил- СоА-производное. Эта реакция формально аналогична двум другим процессам окислительного декарбоксилирования, с которыми мы встречались в гл. 16: окислению пирувата до ацетил- СоА пируватдегидрогеназным комплексом (см. рис. 16-6) и окислению а-кетоглутарата до сукцинил-СоА α-кетоглутаратдегидрогеназным комплексом (с. 194). Все три ферментных комплекса похожи по структуре и осуществляют в сущности один и тот же реакционный механизм. В нем участвуют пять кофакторов (тиаминпирофосфат, FAD, NAD, липоат и кофермент А), а три белка каждого комплекса катализируют гомологичные реакции. Здесь мы имеем случай, когда ферментативные схемы, которые «создавались» для того, чтобы катализировать одну реакцию, были «заимствованы» при дупликации генов, и в дальнейшем адаптировались для осуществления похожих реакций в других путях.
Рис. 18-28. Катаболические пути трех разветвленных аминокислот валина, изолейцина и лейцина. В трех путях, которые существуют во внепеченочных тканях, два первых фермента, как показано здесь, общие. Дегидрогеназный комплекс разветвленных α-кетокислот аналогичен пируватному и α-кетоглутаратному дегидрогеназным комплексам и использует те же пять кофакторов (некоторые здесь не показаны). Дефектный фермент вызывает у людей болезнь кленового сиропа.
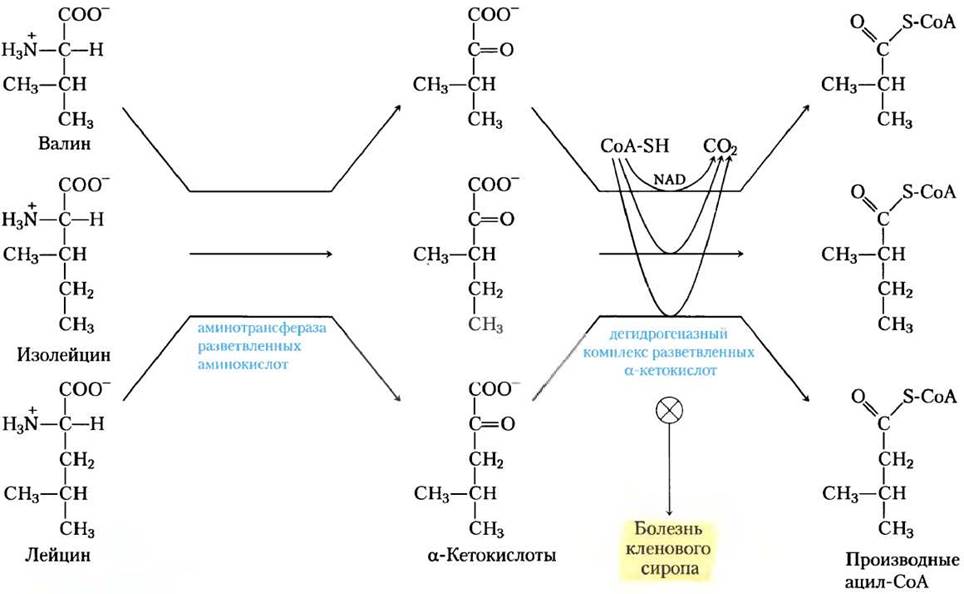
Эксперименты на крысах показали, что дегидрогеназный комплекс разветвленных α-кетокислот регулируется ковалентной модификацией в ответ на содержание разветвленных аминокислот в пище. При низком или неизбыточном содержании разветвленных аминокислот в поглощаемой пище ферментный комплекс фосфорилируется протеинкиназой и таким образом инактивируется. Добавление излишка разветвленных аминокислот в еду вызывает дефосфорилирование и последующую активацию фермента. Обратите внимание, что пируватдегидрогеназный комплекс регулируется похожим образом с помощью фосфорилирования и дефосфорилирования (с. 208).
Есть относительно редкая генетическая болезнь, при которой три разветвленные α-кетокислоты (как и предшествующие им аминокислоты, особенно лейцин) накапливаются в крови и появляются в избытке в моче. Это расстройство названо болезнью кленового сиропа из-за характерного запаха, который придают моче α-кетокислоты, образующиеся дефектным дегидрогеназным комплексом α-кетокислот. Если болезнь не лечить, нарушается развитие мозга, происходит задержка умственного развития и смерть в раннем детстве. Лечение представляет собой жесткий контроль питания, сведение к минимуму, необходимому для нормального роста, потребления валина, изолейцина и лейцина. ■
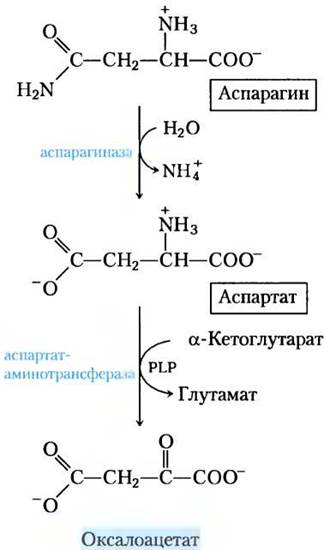
Аспарагин и аспартат расщепляются до оксалоацетата
Углеродные скелеты аспарагина и аспартата в конце концов попадают в цикл трикарбоновых кислот в виде оксалоацетата. Фермент аспарагиназа катализирует гидролиз аспарагина до аспартата, который вступает в реакцию транса- минирования с α-кетоглутаратом с образованием глутамата и оксалоацетата (рис. 18-29).
Рис. 18-29. Катаболический путь аспарагина и аспартата. Обе аминокислоты превращаются в оксалоацетат.
Итак, мы рассмотрели, каким образом 20 протеиногенных аминокислот после потери атомов азота распадаются в реакциях дегидрогенизации, декарбоксилирования и в других реакциях до фрагментов углеродных скелетов шести центральных метаболитов, которые способны поступать в цикл трикарбоновых кислот. Эти компоненты, расщепленные до ацетил-СоА, полностью окисляются до углекислого газа и воды с образованием АТР при окислительном фосфорилировании.
Как и в случае углеводородов и липидов, распад аминокислот в цикле трикарбоновых кислот приводит в итоге к образованию восстановительных эквивалентов (NADH и FADH2). Наш обзор катаболических процессов заканчивается в следующей главе обсуждением клеточного дыхания, в котором эти восстановительные эквиваленты снабжают «топливом» окислительные и энергообразующие процессы в аэробных организмах.
■ В зависимости от конечного продукта деградации некоторые аминокислоты могут быть превращены в кетоновые тела, некоторые — в глюкозу, а некоторые — и в то, и в другое. Таким образом, расщепление аминокислот интегрируется в промежуточный метаболизм и может быть определяющим для выживания при условиях, в которых аминокислоты служат значительным источником метаболической энергии.
■ Углеродные скелеты аминокислот попадают в цикл трикарбоновых кислот в виде пяти интермедиатов: ацетил-СоА, α-кетоглутарата, сукцинил-СоА, фумарата и оксалоацетата. Некоторые также преобразуются в пиру- ват, который может быть превращен либо в ацетил-СоА, либо в оксалоацетат.
■ Аминокислоты, преобразующиеся в пиру- ват, — это аланин, цистеин, глицин, серин, треонин и триптофан. Лейцин, лизин, фенилаланин и триптофан превращаются в ацетил-СоА через ацетоацетил-СоА. Изолейцин, лейцин, треонин и триптофан могут непосредственно превращаться в ацетил-СоА.
■ Аргинин, глутамат, глутамин, гистидин и пролин дают α-кетоглутарат; изолейцин, метионин, треонин и валин образуют сукцинил- СоА; четыре атома фенилаланина и тирозина участвуют в образовании фумарата; аспарагин и аспартат превращаются в оксалоацетат.
Краткое содержание раздела 18.3 Пути деградации углеродного скелета аминокислот
■ После удаления аминогрупп углеродные скелеты аминокислот подвергаются окислению, и далее полученные интермедиаты могут участвовать в цикле трикарбоновых кислот, окисляясь до СО2 и Н2О. В реакциях этих путей участвует ряд кофакторов: тетрагидрофолат и S-аденозилметионин в реакциях переноса одноуглеродных групп и тетрагидробиоптерин в окислении фенилаланина фенилаланиндегидроксилазой.
■ Разветвленные аминокислоты (изолейцин, лейцин и валин), в отличие от других аминокислот, претерпевают распад только во внепеченочных тканях.
■ Ряд серьезных заболеваний человека может быть вызван генетическими дефектами ферментов катаболизма аминокислот.