Биохимия человека Том 1 - Марри Р. 1993
Биоэнергетика и метаболизм углеводов и липидов
Регуляция метаболизма липидов и источники энергии в тканях
Регуляция окисления жирных кислот - Кетогенез
В определенных метаболических условиях, когда происходит быстрое окисление жирных кислот, в печени образуются значительные количества ацетоацетата и D(—)-3-гидрокснбутирата (ß-гидроксибутирата), которые диффундируют в кровь. Ацетоацетат может спонтанно декарбоксилироваться с образованием ацетона. Эти три вещества известны под общим названием кетоновые тела (или ацетоновые тела), иногда их неправильно называют «кетоны»1 (рис. 28.2). Обратимую реакцию превращения ацетоацетата в 3-гидроксибутират катализирует митохондриальный фермент D(—)-3-гидроксибутиратдегидрогеназа; равновесие регулируется отношением [NAD+]/[NADH] в митохондриях, т.е. окислительно-восстановительным статусом. Отношение [3-гидроксибутират]/[ацетоацетат] в крови колеблется между 1:1 и 10:1.
При хорошем питании концентрация кетоновых тел в крови млекопитающих в норме не превышает 1 мг/100 мл (в ацетоновых эквивалентах). У жвачных эта цифра несколько выше из-за образования в стенке рубца 3-гидроксибутирата из масляной кислоты в процессе ферментации. У человека обычно выводится с мочой менее 1 мг кетоновых тел в сутки. Состояние, которое характеризуется повышенным содержанием кетоновых тел в крови или моче, называют соответственно кетонемией (гиперкетонемией) или кетонурией. Общее состояние получило название — кетоз. Ацетоуксусная и 3-гидроксимасляная кислоты являются умеренно сильными кислотами, в крови или тканях они находятся в нейтрализованной форме. Длительное выведение этих кислот вызывает потерю буферных катионов (несмотря на образование аммиака почками), что приводит к истощению щелочного резерва и кетоацидозу. При неконтролируемом сахарном диабете кетоацидоз может иметь фатальные последствия.
1 Не следует пользоваться термином «кетоны крови», поскольку 3-гидроксибутират не является кетоном, кроме того, в крови содержится ряд кетонов, не относящихся к кетоновым телам, например пируват и фруктоза.
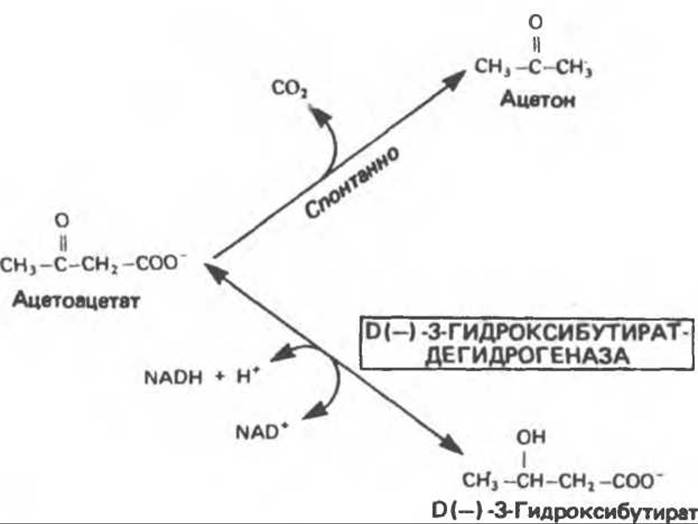
Рис. 28.2. Взаимопревращение кетоновых тел. D(—)-3-Гидроксибутиратдегидрогеназа является митохондриальным ферментом.
Простейшая форма кетоза наблюдается при голодании; при этом происходит исчерпание запасов доступных углеводов, сопряженное с мобилизацией свободных жирных кислот. В качественном плане состояния кетоза, возникающие при различных состояниях, мало различаются. Значительные нарушения метаболизма, приводящие к патологическим состояниям, наблюдаются при диабете, токсикозе беременности у овец и кетозе лактирующих коров. Непатологические формы кетоза наблюдаются при богатой жиром диете и при тяжелых физических нагрузках в период после приема пищи.
У всех животных, кроме жвачных, печень является, по-видимому, единственным органом, поставляющим значительные количества кетоновых тел в кровь. Внепеченочные ткани используют их в качестве субстратов окислительных процессов. Внепеченочные источники кетоновых тел, функционирующие у жвачных при хорошем питании, практически не вызывают у них состояние кетоза.
Поток кетоновых тел из печени во внепеченочные ткани обусловлен функционированием в печени активного ферментативного механизма образования кетоновых тел на фоне очень низкой активности ферментов печени, участвующих в их утилизации. Противоположная ситуация наблюдается во внепеченочных тканях (рис. 28.3).

Рис. 28.3. Образование, утилизация и выведение кетоновых тел. Главный путь показан непрерывными стрелками.
Путь кетогенеза в печени
Ферменты, ответственные за образование кетоновых тел, находятся в основном в митохондриях. Раньше считали, что при окислении молекулы жирной кислоты за счет ее четырех конечных атомов углерода образуется только одна молекула ацетоацетата. Позднее, при объяснении образования более чем одного эквивалента ацетоацетата из одной молекулы длинноцепочечной жирной кислоты, а также образования кетоновых тел из уксусной кислоты, пришли к заключению, что двухуглеродные фрагменты, образующиеся при ß-окислении, конденсируются друг с другом, образуя ацетоацетат. Конденсация происходит путем обращения реакции тиолитического расщепления, в результате 2 молекулы ацетил-СоА образуют ацетоацетил-СоА. Таким образом, ацетоацетил-СоА, являющийся исходным соединением при кетогенезе, образуется либо непосредственно в ходе ß-окисления, либо в результате конденсации ацетил-СоА (рис. 28.4). Предложены два пути образования ацетоацетата из ацетил-СоА. Первый — обычное деацилирование, второй (рис. 28.5) — конденсация молекулы ацетоацетил-СоА с молекулой ацетил-СоА с образованием 3-гидрокси-3-метилглутарил-СоА (ГМГ-СоА), катализируемая 3-гидрокси-3-метилглутарил-СоА-синтазой. Под действием другого фермента, локализованного в митохондриях. 3-гидрокси-3-метилглутарил-СоА-лиазы, ацетил-СоА отщепляется от ГМГ-СоА, и образуется свободный ацетоацетат. Атомы углерода отщепившейся молекулы ацетил-СоА исходно принадлежали молекуле ацетоацетил-СоА (рис. 28.5). Для осуществления кетогенеза необходимо, чтобы в митохондриях находились оба фермента (такое сочетание ферментов имеется только в печени и эпителии рубца).
В настоящее время доминирует представление, согласно которому образование кетоновых тел происходит главным образом по ГМГ-СоА-пути. Хотя при голодании наблюдается значительная активация ГМГ-СоА-лиазы, имеющиеся данные не свидетельствуют о том, что этот фермент лимитирует скорость кетогенеза.
Ацетоацетат может превращаться в D(—)-3-гидроксибутират под действием D(—)-3-гидроксибутиратденидрогеназы, находящейся во многих тканях, в том числе в печени. В количественном отношении D(—)-3-гидроксибутират является при кетозе доминирующим кетоновым телом в крови и моче.
Утилизация кетоновых тел во внепеченочных тканях
В печени функционирует активный механизм образования ацетоацетата из ацетоацетил-СоА. Активация образовавшегося ацетоацетата может произойти только в цитозоле, где он является предшественником при синтезе холестерола, однако активность этого пути сравнительно невелика; в результате в печени происходит образование избытка кетоновых тел.
Во внепеченочных тканях протекают две реакции, в результате которых ацетоацетат активируется в ацетоацетил-СоА. Одна из них протекает с участием сукцинил-СоА и катализируется сункцннил-СоА-ацетоацетат-СоА-трансферазой. Ацетоацетат реагирует с сукцинил-СоА, при этом СоА переносится на ацетоацетат и образуются ацетоацетил-СоА и сукцинат.

Другая реакция осуществляется путем активирования ацетоацетата АТР в присутствии СоА, она катализируется ацетоацетил-СоА-синтетазой.

D(—)-3-Гидроксибутират может активироваться синтетазой во внепеченочных тканях; однако доминирующим путем является превращение в ацетоацетат, катализируемое D(—)-3-гидроксибутиратдегидрогеназой при участии NAD+, и последующее активирование с образованием ацетоацетил-СоА. Ацетоацетил-СоА, образовавшийся в результате этих реакций, расщепляется при участии тиолазы до ацетил-СоА; последний окисляется в цикле лимонной кислоты (рис. 28.4).
Кетоновые тела окисляются во внепеченочных тканях пропорционально их содержанию в крови; они подвергаются окислению предпочтительно по сравнению с глюкозой и СЖК. При повышении содержания кетоновых тел в крови их окисление усиливается до тех пор, пока при концентрации 70 мг/100 мл они не насыщают окислительный механизм. В этом состоянии, по-видимому, большая часть кислорода, потребляемого животными, расходуется на окисление кетоновых тел.
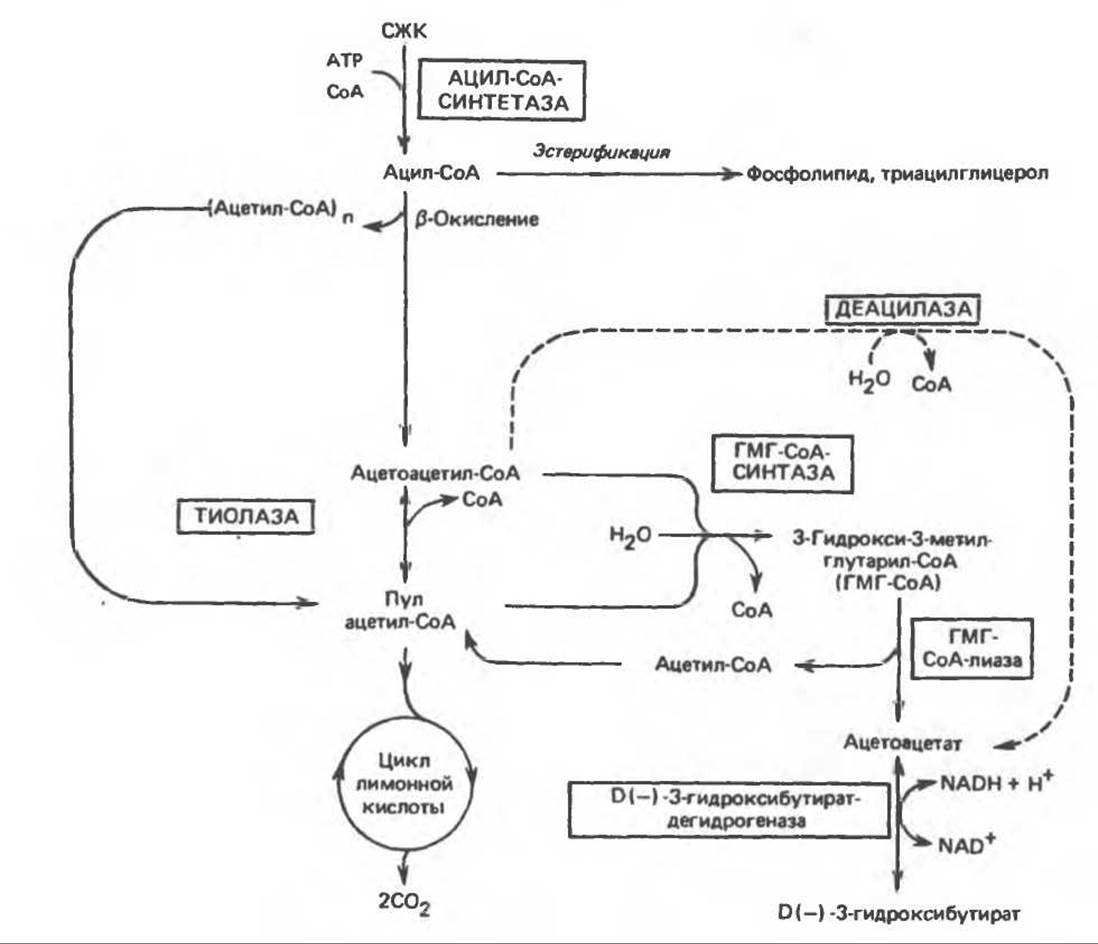
Рис. 28.4. Путь кетогенеза в печени. СЖК — свободные жирные кислоты: ГМГ — 3-гидрокси-3-метилглутарил.
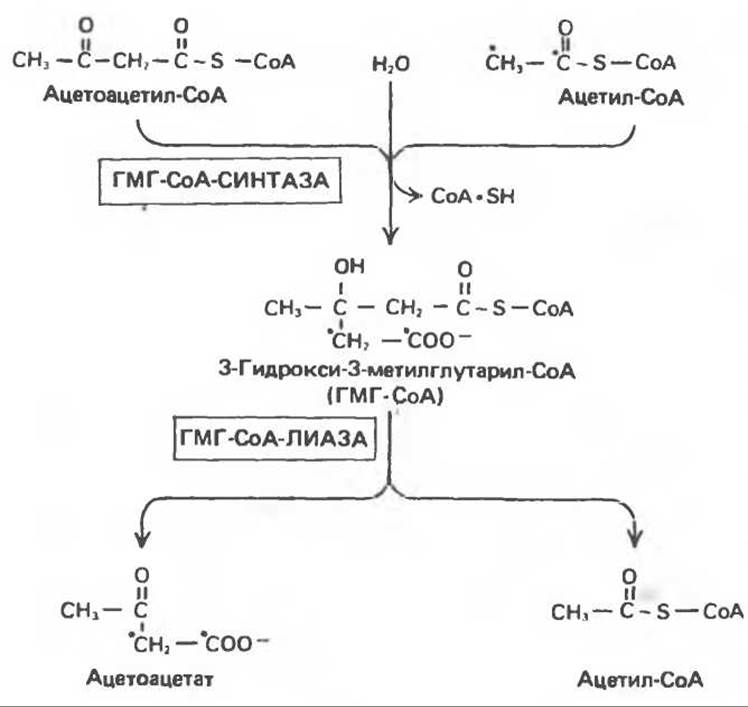
Рис. 28.5. Образование ацетоацетата из ацетоацетил-СоА (на промежуточной стадии образуется ГМГ-СоА).
Большинство данных свидетельствует о том, что причиной кетонемии является увеличение образования кетоновых тел в печени, а не недостаточная их утилизация во внепеченочных тканях. В то же время результаты экспериментов с крысам», у которых была удалена поджелудочная железа, показывают, что при тяжелой форме диабета кетоз может усиливаться в результате пониженной способности организма к катаболизму кетоновых тел. При умеренной кетонемии с мочой выводится только несколько процентов от общего количества образующихся кетоновых тел.
Поскольку при экскреции кетоновых тел почками наблюдаются порогоподобные эффекты (не являющиеся, однако, истинными пороговыми эффектами), которые варьируют у видов и отдельных особей, о тяжести кетогенеза следует судить по уровню кетоновых тел в крови, а не в моче.
Ацетоацетат и D(—)-3-гидроксибутират легко окисляются во внепеченочных тканях, а окисление ацетона in vivo затруднено.
Регуляция кетогенеза
Выделяют три стадии, на которых соответствующие факторы могут осуществлять регуляцию кетогенеза. (1) Кетоз не возникает in vivo до тех пор, пока не происходит увеличения уровня свободных жирных кислот в крови, образующихся в результате липолиза триацилглицерола в жировой ткани. Жирные кислоты являются предшественниками кетоновых тел в печени. Как у сытых, так и у голодных животных печень обладает способностью поглощать до 30% и более свободных жирных кислот, проходящих через нее, поэтому при высоких концентрациях этих кислот поглощение их довольно значительно. Следовательно, для регуляции кетогенеза важны факторы, контролирующие стадию мобилизации свободных жирных кислот из жировой ткани (рис. 28.6). (2) Возможны два пути превращения свободных жирных кислот после их поступления в печень и перехода в активные ацил-СоА-производные, а именно эстерификация с образованием преимущественно триацилглицеролов и фосфолипидов и ß-окисление до ацетил-СоА. (3) В свою очередь ацетил-СоА может либо окисляться в цикле лимонной кислоты, либо вступать на путь кетогенеза, образуя кетоновые тела.
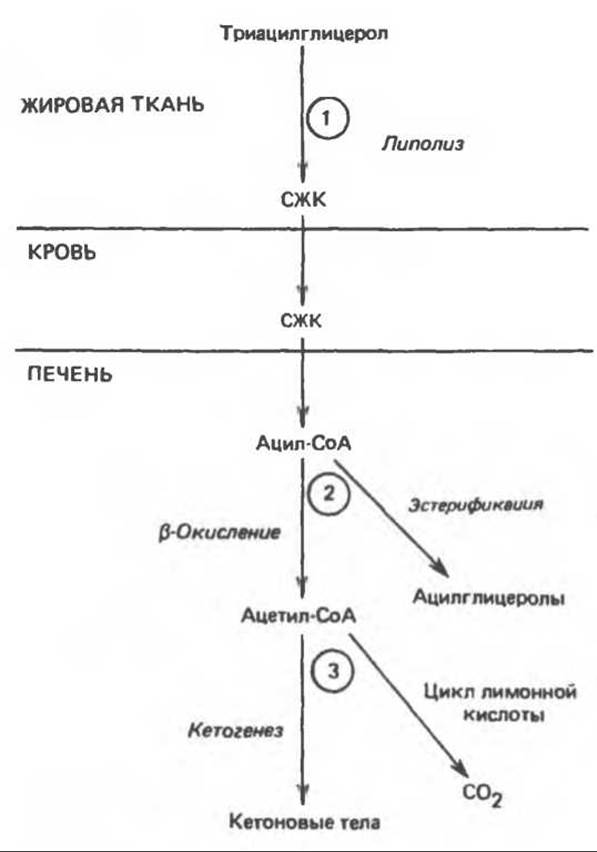
Рис. 28.6. Регуляция кетогенеза. 1—3 — три ключевые стадии на пути метаболизма свободных жирных кислот (СЖК), которые определяют количество образующихся кетоновых тел.
Одним из возможных антикетогенных факторов регуляции является эстерификация свободных жирных кислот, которая зависит от наличия в печени предшественников, обеспечивающих образование достаточного количества глицерол-3-фосфата. Однако в опытах с перфузируемой печенью, взятой у голодного животного, доступность глицерол-3-фосфата не лимитировала эстерификацию. Не вполне ясно, всегда ли доступность глицерол-3-фосфата в печени лимитирует скорость эстерификации; не имеется также убедительных данных о том, что in vivo скорость эстерификации лимитирует активность соответствующих ферментов. Вряд ли это имеет место, поскольку в печени не накапливаются ни свободные жирные кислоты, ни промежуточные продукты их эстерификации на пути образования триацилглицерола (рис. 25.1). Активность фосфатидатфосфогидролазы в печени возрастает в условиях избыточного синтеза триацилглицеролов.
В опытах с перфузируемой печенью было показано, что в печени сытых крыс эстерифицируется значительно большее количество 14С-свободных жирных кислот, чем в печени голодных животных, у последних соответствующая часть неэстерифицированных жирных кислот окисляется до 14СО2 или 14С- кетоновых тел. Эти результаты можно объяснить тем, что поступление длинноцепочечных ацильных групп внутрь митохондрий, где происходит ß-окисление, регулируется карнитин-пальмитоилтрансферазой I, локализованной в митохондриальной мембране (рис. 23.1); данный фермент малоактивен у сытых животных, у которых окисление жирных кислот заторможено, и весьма активен при голодании, которое сопровождается усиленным окислением жирных кислот. Макгарри и Фостер (McGarry, Foster, 1980) показали, что малонил-СоА — исходный интермедиат в синтезе жирных кислот (см. рис. 23.5) (его концентрация увеличивается в сытом состоянии) ингибирует карнитин-пальмитоилтрансферазу и выключает ß-окисление. Таким образом, в сытом состоянии происходит активный липогенез и достигается высокая концентрация малонил-СоА, ингибирующего карнитин-пальмитоилтрансферазу I (рис. 28.7). Если концентрация свободных жирных кислот в клетках печени невелика, они почти полностью превращаются путем эстерификации в ацилглицеролы и выводятся из печени в составе ЛПОНП. Однако в начале голодания, когда концентрация свободных жирных кислот возрастает, ацетил-СоА-карбоксилаза ингибируется, а концентрация малонил-СоА уменьшается, ингибирование карнитин-пальмитоилтрансферазы прекращается и создаются условия для усиленного окисления ацил-СоА. Эти процессы усиливаются при голодании в результате уменьшения отношения [инсулин]/[глюкагон], что вызывает ускорение липолиза и высвобождение свободных жирных кислот в жировой ткани и ингибирование ацетил-СоА-карбоксилазы в печени.
При повышении уровня свободных жирных кислот в сыворотке крови пропорционально больше свободных жирных кислот превращается в кетоновые тела и соответственно меньше окисляется в цикле лимонной кислоты до СО2. При этом в результате регулирования достигается такое распределение ацетил-СоА между путем кетогенеза и путем окисления до СО2, что свободная энергия, запасаемая в форме АТР в процессе окисления свободных жирных кислот, остается постоянной. При полном окислении 1 моля пальмитата путем ß-окисления и последующего образования СО2 в цикле лимонной кислоты генерируется 129 молей АТР (см. гл. 23); если же конечным продуктом является ацетоацетат, образуется всего 33 моля АТР, а если 3-гидроксибутират — то только 21 моль. Следовательно, кетогенез можно рассматривать как механизм, позволяющий печени окислять большие количества жирных кислот, используя реакции, входящие в систему окислительного фосфорилирования (при этом генерация макроэргов невелика).
Предложен ряд других гипотез по поводу переключения пути окисления жирных кислот в направлении кетогенеза. Теоретически снижение концентрации оксалоацетата в митохондриях должно понижать возможность метаболизма ацетил-СоА в цикле лимонной кислоты. Причиной уменьшения концентрации оксалоацетата может являться увеличение отношения [NADH]/[NAD+] при усиленном ß-окислении. Кребс предположил, что, поскольку оксалоацетат находится также на главном пути глюконеогенеза, усиление этого процесса, ведущее к снижению уровня оксалоацетата, может являться причиной тяжелых форм кетоза, в частности при диабете и кетозе крупного рогатого скота. Уттер и Кич (Utter, Keech) показали, что пируваткарбоксилаза, катализирующая превращение пирувата в оксалоацетат, активируется ацетил-СоА. Следовательно, при значительном количестве ацетил-СоА необходимо достаточное для запуска реакции конденсации в цикле лимонной кислоты количество оксалоацетата.
В заключение суммируем, что кетоз возникает в результате недостатка доступных углеводов, это обстоятельство следующим образом способствует кетогенезу (рис. 28.6 и 28.7). (1) Оно приводит к дисбалансу между эстерификацией и липолизом в жировой ткани, в результате которого свободные жирные кислоты поступают в кровоток. Эти кислоты являются главным субстратом для образования кетоновых тел в печени; поэтому все факторы как метаболические, так и эндокринные, влияющие на высвобождение свободных жирных кислот из жировой ткани, воздействуют также на процесс кетогенеза. (2) После поступления свободных жирных кислот в печень распределение их по путям эстерификации и окисления регулируется карнитин-пальмитоилтрансферазой I, активность которой зависит (опосредованно) от концентрации свободных жирных кислот и гормонального статуса печени. (3) При увеличении количества окисляемых жирных кислот возрастает доля образуемых кетоновых тел и соответственно уменьшается доля субстрата, который подвергается катаболизму до СО2; при этом в результате регуляции общий выход АТР остается постоянным.
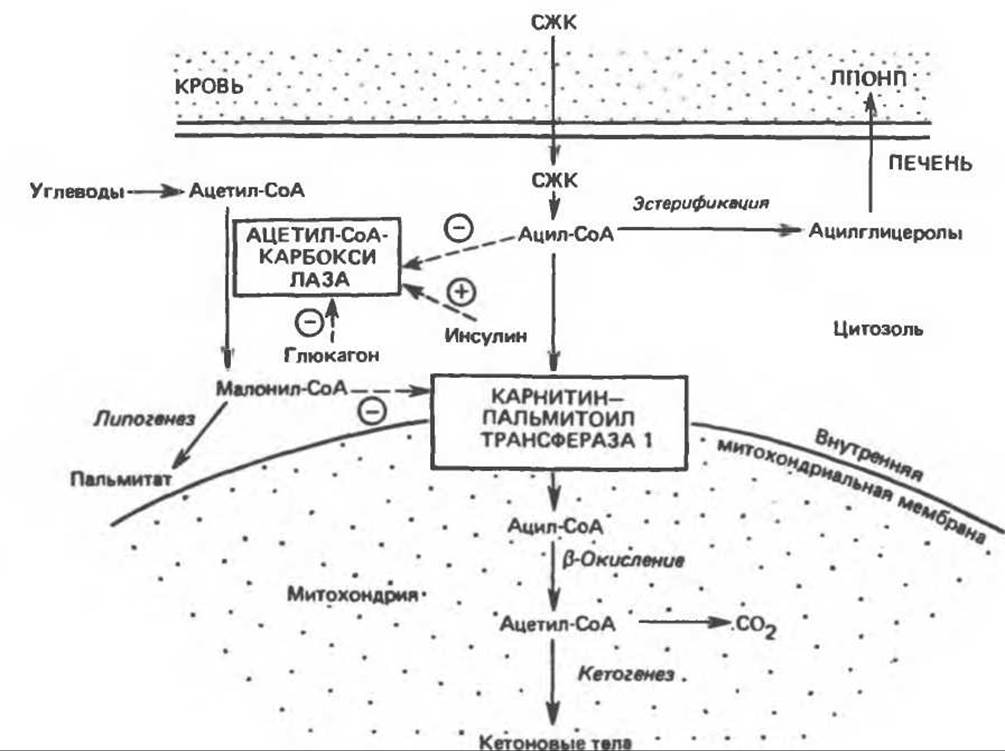
Рис. 28.7. Регуляция окисления длинноцепочечных жирных кислот в печени. СЖК — свободные жирные кислоты, ЛПОНП — липопротеины очень низкой плотности. Пунктирными линиями показаны положительные (⊕) и отрицательные (⊝) регуляторные эффекты, а сплошными линиями — поток субстрата.
Кетоз in vivo
Наблюдаемый при голодании и при потреблении жирной пищи кетоз протекает в относительно легкой форме по сравнению с кетозом, развивающимся при неконтролируемом диабете, токсикозе беременности у овец или у крупного рогатого скота в период лактации. Основная причина этого, по-видимому, заключается в том, что при указанных заболеваниях количество доступных для тканей углеводов существенно меньше, чем при голодании и потреблении жирной пищи. Так, при умеренном диабете, приеме жирной пищи и хроническом голодании в печени сохраняется гликоген (количество его варьирует); менее выражено снижение уровня свободных жирных кислот. Этим, вероятно, можно объяснить менее тяжелую форму кетоза, которая наблюдается в этих случаях. У жвачных кетоз протекает на фоне значительного снижения содержания глюкозы в крови, что связано с обеспечением потребностей плода или молочных желез при интенсивной лактации (рис. 28.8). В результате у жвачных может развиться острая гипогликемия, при которой в печени практически не остается гликогена. В этих условиях кетоз протекает в тяжелой форме. По мере развития гипогликемии уменьшается секреция инсулина, в результате не только снижается утилизация глюкозы, но и усиливается липолиз в жировой ткани.
При сахарном диабете недостаток или отсутствие инсулина влияют, вероятно, в первую очередь на метаболизм в жировой ткани, крайне чувствительной к этому гормону. В результате высвобождения большого количества свободных жирных кислот их уровень в плазме крови может оказаться в два с лишним раза выше, чем у здорового голодного человека. Наблюдается также изменение активности ряда ферментов в печени, в результате увеличивается скорость глюконеогенеза и скорость поступления глюкозы в кровь (несмотря на высокую концентрацию глюкозы в крови).