Биохимия человека Том 1 - Марри Р. 1993
Метаболизм белков и аминокислот
Катаболизм углеродного скелета аминокислот
Аминокислоты, образующие ацетил-кофермент А
Все аминокислоты, образующие пируват (аланин, цистеин, цистин, глицин, гидроксипролин, серин и треонин), могут превращаться в ацетил-СоА. Кроме того, 5 аминокислот образуют ацетил-СоА без промежуточного образования пирувата. К числу этих аминокислот относятся ароматические аминокислоты фенилаланин, тирозин и триптофан, основная аминокислота лизин и нейтральная аминокислота с разветвленной цепью лейцин.
Тирозин
А. Общая последовательность реакций. Пять последовательных ферментативных реакций превращают тирозин в фумарат и ацетоацетат (рис. 31.13): 1) реакция переаминирования с образованием n-гидроксифенилпирувата; 2) окисление с миграцией трехуглеродной боковой цепи и декарбоксилирование, приводящее к образованию гомогентизата; 3) окисление гомогентизата до малеилацетоацетата; 4) изомеризация малеилацетоацетата в фумарилацетоацетат и 5) гидролиз фумарилацетоацетата с образованием фумарата и ацетоацетата. Ацетоацетат может далее подвергнуться тиолитическому расщеплению на ацетат и ацетил-СоА.
Несколько интермедиатов метаболизма тирозина было идентифицировано при изучении генетического заболевания человека алкаптонурии. Больные алкаптонурией экскретируют с мочой гомогентизат и немало полезной информации было получено при добавлении в пищу пациентов предполагаемых предшественников гомогентизата.

Рис. 31.12. Интермедиаты катаболизма а- гидроксипролина в тканях млекопитающих. аКК — а-кетокислота, а-АК — а-аминокислота. Цифры в кружочках указывают вероятные места метаболических нарушений при: 1 — гипергидроксипролинемии, 2 — гиперпролинемии типа II.
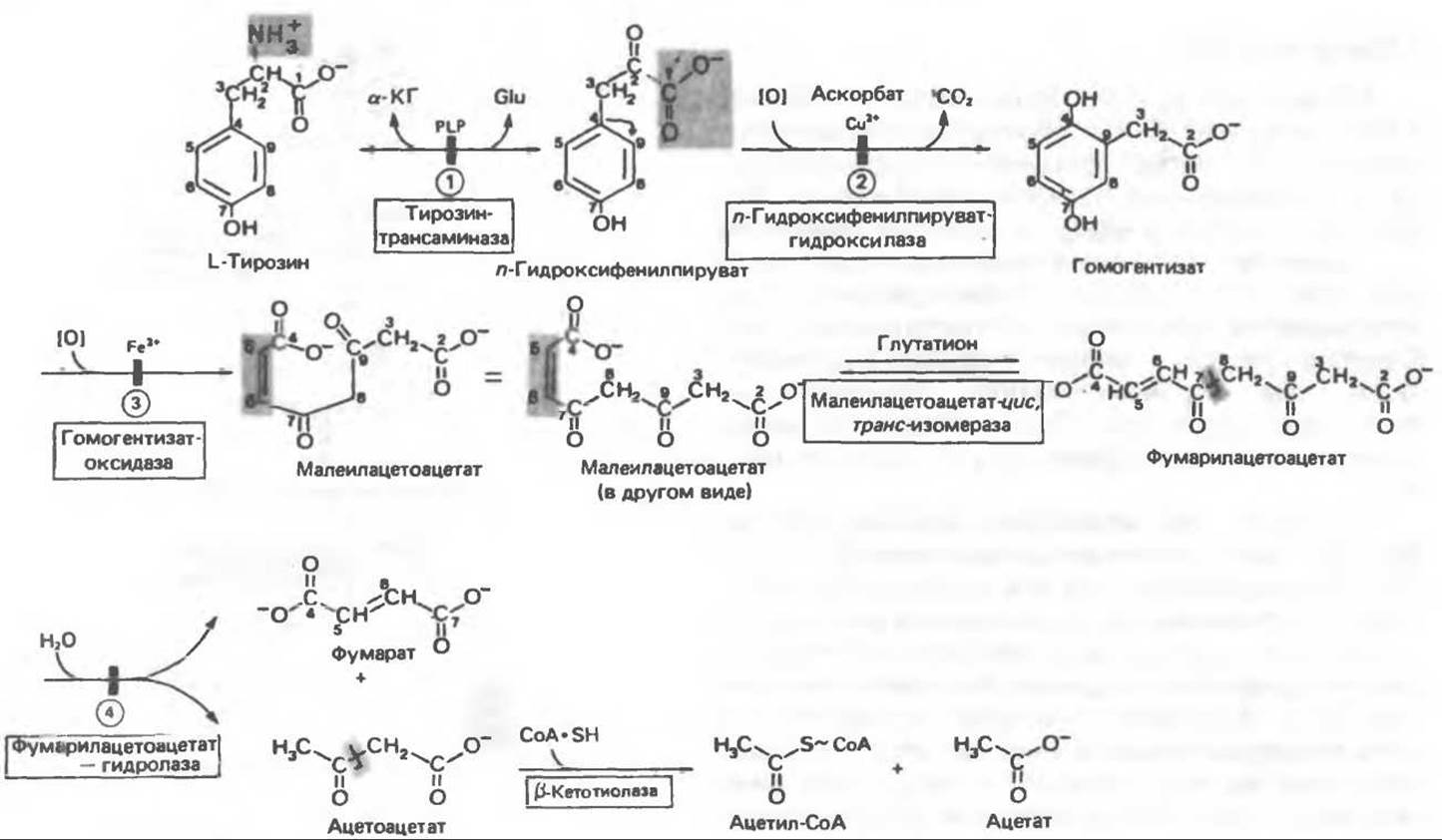
Рис. 31.13. Интермедиаты катаболизма тирозина. За исключением реакции, катализируемой ß-кетотиолазой, все остальные реакции обсуждаются в тексте. Чтобы читателю было легче проследить за судьбой каждого атома углерода, они пронумерованы (см. также рис. 31.15). а-КГ — а-кетоглутарат, Glu — глутамат, PLP — пиридоксальфосфат. Цифры в кружочках указывают вероятные места метаболических нарушений при 1 — тирозинемии типа II, 2 — тирозинемии новорожденных, 3 — алкаптонурии и 4 — тирозинемии типа I, или тирозинозе.
Б. Переаминированне тирозина. Переаминирование тирозина с образованием n-гидроксифенилпирувата катализируется тирозин-а-кетоглутараттрансаминазой — индуцируемым ферментом печени млекопитающих.
В. Окисление n-гидрокснфеннлпнрувата в гомогентизат. Хотя реакция (рис. 31.13) выглядит как гидроксилирование n-гидроксифенилпирувата, сопровождающееся окислительным удалением карбоксильного углерода, в действительности сочетанно происходит миграция боковой цепи и гидроксилирование кольца. n-Гидроксифенилпируват-гидролаза является медьсодержащим металлопротеином, сходным с тирозиназой. Хотя in vitro кофактором реакции могут служить не только аскорбат, но и другие восстанавливающие агенты, больные цингой экскретируют с мочой неполностью окисленные продукты метаболизма тирозина.
Г. Превращение гомогентизата в фумарат и ацетоацетат. Окислительная реакция, приводящая к разрыву бензольного кольца гомогентизата и образованию малоилацетоацетата, катализируется гомогентизат-оксидазой — железосодержащим металлопротеином печени млекопитающих.
Превращение малеилацетоацетата в фумарилацетоацетат является цис-транс-изомеризацией относительно двойной связи и катализируется малеилацетоацетат-цис,транс-изомеразой — (—SH)-зависимым ферментом печени млекопитающих. Гидролиз фумарилацетоацетата, катализируемый фумарилацетоацетат-гидролазой, приводит к образованию фумарата и ацетоацетата. Последний затем может быть превращен в ацетил-СоА и ацетат в реакции, катализируемой ß-кетотиолазой (см. гл. 23).
Нарушения катаболизма тирозина. Тяжелые нарушения катаболизма приводят к тирозинемии, тирозинурии и фенолацидурии.
А. Тирозинемия типа I (тирозиноз). Тирозиноз характеризуется накоплением метаболитов, снижающих активность ряда ферментов и транспортных систем. Патофизиология этого нарушения является, следовательно, весьма сложной. Дефектными ферментами являются, вероятно, фумарилацетоацетат-гидролаза (рис. 31.13) и малеилацетоацетат- гидролаза.
Известны острая и хроническая формы тирозиноза. Острая форма характерна для младенческого возраста, ее признаками являются понос, рвота, «капустный» запах, задержка в развитии. Если не проводится лечение, летальный исход наступает в возрасте 6—8 месяцев из-за недостаточности печени. Хроническая тирозинемия характеризуется сходными, но более умеренно выраженными симптомами; летальный исход наступает в возрасте примерно 10 лет. Содержание тирозина в плазме повышается до 6—12 мг/100 мл, повышено содержание и некоторых других аминокислот, особенно метионина. Лечение включает диету с пониженным содержанием тирозина и фенилаланина, а в ряде случаев также и метионина.
Б. Тирозинемия типа II (синдром Рихнера—Ханхарта). Предполагаемой причиной метаболических нарушений при тирозинемии типа II является недостаточность тирозин-трансаминазы печени (рис. 31.13). Клинические проявления включают повышение содержания тирозина в плазме (4—5 мг/100 мл), характерные поражения глаз и кожи и умеренную умственную отсталость. Отмечались также случаи членовредительства, нарушения тонкой координации движений. Тирозин является единственной аминокислотой, концентрация которой в моче повышена. Тем не менее клубочковая фильтрация и обратное всасывание тирозина остаются в пределах нормы. В число метаболитов, экскретируемых с мочой, входят n-гидроксифенилпируват, n-гидроксифениллактат, n-гидроксифенилацетат, N-ацетилтирозин и тирамин (рис. 31.14).
В. Тирозинемия новорожденных. Причиной болезни считается недостаточность n-гидроксифенилпируват-гидроксилазы (рис. 31.13). Повышено содержание в крови тирозина и фенилаланина, а в моче содержание тирозина, n-гидроксифенилацетата, N-ацетилтирозина и тирамина. При лечении назначают бедную белком диету.
Г. Алкаптонурия. Это наследственное метаболическое нарушение описано в медицинской литературе еще в XVI в., оно было охарактеризовано в 1859 г. Болезнь представляет значительный исторический интерес, поскольку именно на основе ее изучения Гаррод выдвинул идею о наследственных метаболических нарушениях. Наиболее ярким клиническим проявлением этой болезни является потемнение мочи при условиях доступа воздуха. На более поздних стадиях заболевания наблюдается общая пигментация соединительной ткани (охроноз) и развивается артрит. Причиной болезни является недостаточность гомогентизат-оксидазы (рис. 31.13). Субстрат фермента, гомогентизат, экскретируется с мочой; при окислении на воздухе он образует темнокоричневый пигмент. Зарегистрировано свыше 600 случаев заболевания, частота заболевания оценивается в 2—5 случаев на миллион новорожденных.

Рис. 31.14. Альтернативные катаболиты тирозина. n-Гидроксифенилацетальдегид является интермедиатом стадии окисления тирамина в n-гидроксифенилацетат.
Алкаптонурия наследуется по аутосомно-рецессивному типу. В настоящее время не найдено диагностических методов выявления гетерозигот. Хотя механизм охроноза не установлен, полагают, что он обусловлен окислением гомогентизата полифенолоксидазой, приводящим к образованию бензохиноацетата, который далее полимеризуется и связывается с макромолекулами соединительной ткани.

Фенилаланин
Фенилаланин сначала превращается в тирозин при участии фенилаланингидроксилазы (см. рис. 29.11). Распределение изотопной метки в амфиболических продуктах фумарате и ацетоацетате (рис. 31.15) такое же, как и в соответствующих продуктах катаболизма тирозина (рис. 31.13).
Метаболические нарушения катаболизма фенилаланина. Главные метаболические нарушения состоят в блокировании превращения фенилаланина в тирозин (см. рис. 29.12). Можно выделить три причины нарушений: недостаточность фенилаланингидроксилазы (гиперфенилаланинемия типа I, или классическая фенилкетонурия), недостаточность дигидробиоптеринредуктазы (гиперфенилаланинемия типов II и III) и нарушения биосинтеза дигидробиоптерина (гиперфенилаланинемия типов IV и V). Были также зарегистрированы нарушения других типов (табл. 31.3).
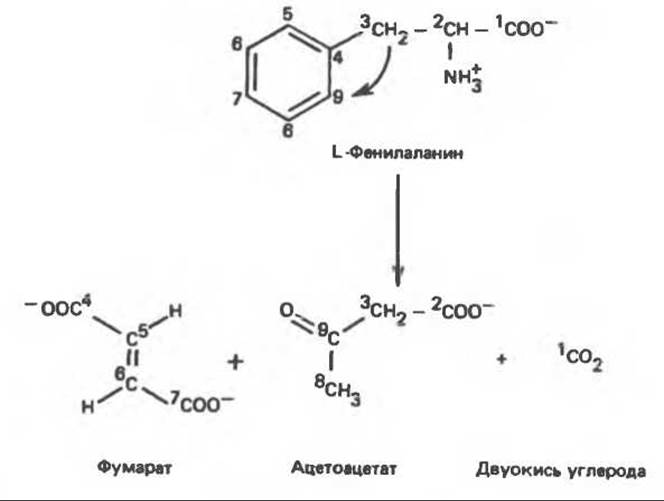
Рис. 31.15. Катаболическая судьба атомов углерода фенилаланина. Распределение изотопной метки в конечных катаболитах фенилаланина (и тирозина).
Главным последствием гиперфенилаланинемии типа 1 (классическая фенилкетонурия, ФКУ) при отсутствии лечения является умственная отсталость (ниже 70 баллов по стандартному тесту для детей старшего возраста). Дополнительными клиническими симптомами являются припадки, психозы, экзема и «мышиный» запах. При раннем диагнозе и своевременном лечении эти симптомы могут не развиться. В связи с наличием экспериментальных моделей заболевания, а также благодаря возможности путем регулирования диеты предотвращать неизбежное в другом случае состояние, характеризующееся умственной отсталостью, ФКУ служит моделью для изучения этого состояния при других метаболических нарушениях. При классической ФКУ, которая является наследственным нарушением, встречающимся с частотой 1 на 10000 новорожденных, содержание в печени компонента 1 фенилаланингидроксилазы (см. рис. 29.12) составляет около 25% от нормы; при этом фермент оказывается нечувствительным к регуляторному действию фенилаланина.
Организм пациента оказывается не способным превращать фенилаланин в тирозин, в результате образуются альтернативные катаболиты фенилаланина (рис. 31.16). В их число входят фенилпировиноградная кислота (продукт дезаминирования фенилаланина), фенилмолочная кислота (продукт восстановления фенилпировиноградной кислоты) и фенилуксусная кислота, образующаяся путем декарбоксилирования и окисления фенилпировиноградной кислоты. Большая часть фенилацетата в печени конъюгирует с глутамином и экскретируется с мочой в виде конъюгата фенилацетилглутамина. В табл. 31.4 приведены концентрации метаболитов фенилаланина в крови и моче у пациентов с фенилкетонурией. Присутствие в моче кетокислоты фенилпирувата определило и само название болезни — фенилкетонурия.
Таблица 31.3. Типы гиперфенилаланинемии 1)
|
Тип |
Название болезни |
Причина болезни |
Метод лечения |
|
I |
Фенилкетонурия |
Отсутствие Phe-гидроксилазы |
Диета с пониженным содержанием Рhе |
|
11 |
Устойчивая гиперфенилаланинемия |
Снижение уровня Phe-гидроксилазы |
Наблюдение или периодическая диетотерапия |
|
111 |
Кратковременная легкая гиперфенилаланинемия |
Недостаток гидроксилазы в период развития |
Тот же, что и в предыдущем случае |
|
IV |
Недостаточность дигидроптеридин-редуктазы |
Отсутствие или недостаток дигидроптеридин-редуктазы |
ДОФА, 5-гидрокситриптофан, карби-ДОФА |
|
V |
Ненормальная функция дигидробиоптерина |
Нарушения в системе синтеза дигидробиоптерина |
ДОФА, 5-гидрокситриптофан, карби-ДОФА |
|
VI |
Устойчивая гиперфенилаланинемия и тирозинемия |
Катаболизм тирозина |
Диета с низким содержанием Phe |
|
VII |
Кратковременная тирозинемия у новорожденных |
Ингибирование n-гидроксифенилпируват-оксидазы |
Витамин С |
|
VIII |
Наследственная тирозинемия |
Недостаточная активность 1. n-гидроксифенилпируват-диоксигеназы 2. цитоплазматической тирозина-минотрансферазы |
Диета с низким содержанием Туr |
|
Недостаток фумарилацетоацетата |
Диета с низким содержанием Туг плюс инъекции глутатиона |
1) Данные взяты с некоторыми изменениями из статьии Tourian A., Sidbury J. В.: Phenylketonuria and hyperphenylaninemia (р. 273) in: Stanbury J. В. et al. (eds): The Metabolic Basis of Inherited Disease, 5th ed. McGraw-Hill, 1983.
При блокировании нормальных путей катаболизма фенилаланина на первый план выходят несколько катаболических реакций, которые протекают в нормальной печени, но обычно имеют второстепенное значение. У больных фенилкетонурией в крови и моче появляются фенилпируват, фениллактат, фенилацетат и фенилацетилглутамин (рис. 31.16). Хотя присутствие фенилпирувата в моче больных фенилкетонурией может быть установлено путем простого биохимического анализа, для убедительного диагноза необходимо зарегистрировать повышение содержания фенилаланина в плазме крови.
Прогрессирующее развитие нарушений умственного развития у детей, больных фенилкетонурией, можно предотвратить путем назначения диеты с очень низким содержанием фенилаланина. По достижении шестилетнего возраста эту диету можно отменить, поскольку к этому времени повышенные концентрации фенилаланина и его производных перестают оказывать неблагоприятное действие на мозг.
Содержание фенилаланина в плазме можно измерить с помощью автоматизированного микрометода, для которого требуются пробы крови не более 20 мкл. Следует отметить, что ненормально высокое содержание фенилаланина у детей, больных фенилкетонурией, обнаруживается лишь на третий или четвертый день жизни, поскольку они потребляют незначительное количество пищевого белка. Далее, у недоношенных детей можно получить ложную положительную реакцию в связи с замедленным образованием ряда ферментов метаболизма фенилаланина. Полезным, но не вполне надежным массовым тестом может служить обнаружение повышенного содержания фенилпирувата с моче с использованием в качестве реагента хлорида железа.
Таблица 31.4. Метаболиты фенилаланина, накапливающиеся в плазме и моче больных фенилкетонурией
|
Метаболит |
Содержание в плазме нг/100 мл) |
Содержание в моче (мг/100 мл) |
||
|
в норме |
при фенилкетонурии |
в норме |
при фенилкетонурии |
|
|
Фенилаланин |
1—2 |
15—63 |
30 |
300—1000 |
|
Фенилпируват |
0,3—1,8 |
300—2000 |
||
|
Фениллактат |
290—550 |
|||
|
Фенилацетат |
Повышенное содержание |
|||
|
Фенилацетилглутамин |
200—300 |
2400 |
||
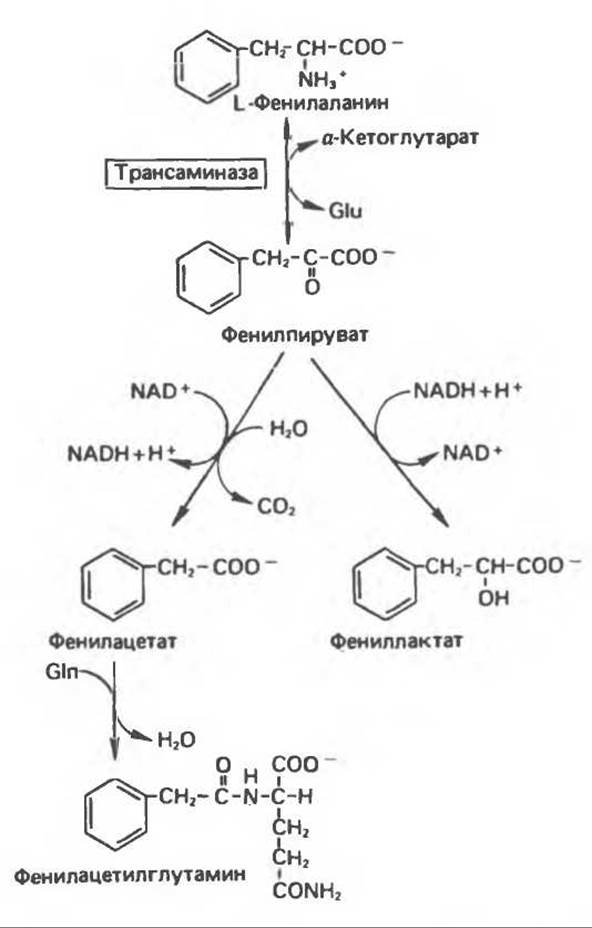
Рис. 31.16. Альтернативные пути катаболизма фенилаланина, имеющие особое значение при фенилкетонурии. Указанные реакции протекают также и в печени здоровых людей, но при нормальном функционировании фенилаланин-гидроксилазы они не имеют существенного значения. Glu — глутамат, Gln — глутамин.
После введения фенилаланина пациентам с фенилкетонурией содержание этой аминокислоты в крови долгое время остается высоким, что свидетельствует о снижении толерантности к фенилаланину. Вместе с тем ненормально низкая толерантность к введенному фенилаланину и повышенное содержание последнего в период между приемами пищи характерно также и для родителей, больных фенилкетонурией. Таким образом, дефектный ген, ответственный за фенилкетонурию, может быть выявлен и у фенотипически нормальных гетерозиготных родителей.
Лизин
Лизин составляет исключение из общего правила, согласно которому первой стадией катаболизма аминокислоты является удаление а-аминогруппы путем переаминирования. В тканях млекопитающих ни а-, ни ε-аминогруппы L-лизина не участвуют в переаминировании. В организме млекопитающих углеродный скелет L-лизина переходит в состав а- аминоадипата и а-кетоадипата (рис. 31.17). Первоначально полагали, что деградация L-лизина идет через стадию образования пипеколиновой кислоты — циклической иминокислоты. В печени D-лизин действительно превращается в пипеколат, однако распад L-лизина происходит через стадию образовав ния сахаропина (рис. 31.18) — интермедиата биосинтеза лизина у грибов.
L-Лизин сначала конденсируется с а-кeтоглутаратом, при этом отщепляется молекула воды и образуется шиффово основание. Далее происходит восстановление этого соединения в сахаропин при участии соответствующей дегидрогеназы (рис. 31.18), а затем окисление сахаропина другой дегидрогеназой. Расщепление продукта водой приводит к образованию L-глутамата и L-a-аминоадипат-δ-полуальдегида. Суммарный эффект этой серии реакций эквивалентен удалению ε-аминогруппы лизина путем переаминирования; L-лизин и а-кетоглутарат превращаются в а-аминоадипат-δ-полуальдегид и глутамат. В качестве кофакторов в процессе участвуют NAD+ и NADH, при этом в итоге не происходит ни окисления, ни восстановления.
В ходе дальнейшего катаболизма а-аминоадипат путем переаминирования превращается в а-кетоадипат, после чего, вероятно, происходит окислительное декарбоксилирование а-кетоадипата с образованием глутарил-СоА. Лизин является одновременно и гликогенной, и кетогенной аминокислотой; природа же катаболитов глутарил-СоА, образующихся в организме млекопитающих, не установлена.
Метаболические нарушения катаболизма лизина. Описаны два редких метаболических нарушения катаболизма лизина. Оба они являются следствием дефектности ферментов, осуществляющих катаболизм лизина до ацетоацетил-СоА, и в обоих случаях первичное нарушение, вероятно, блокирует превращение L-лизина и а-кетоглутарата в сахаропин (рис. 31.18).
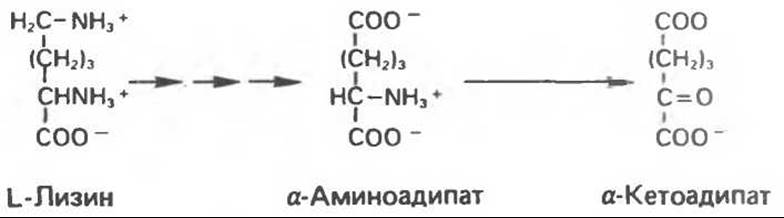
Рис. 31.17. Превращение L-лизина в a-аминоадипат и a-кетоадипат. Несколько стрелок подряд означают промежуточные реакции.
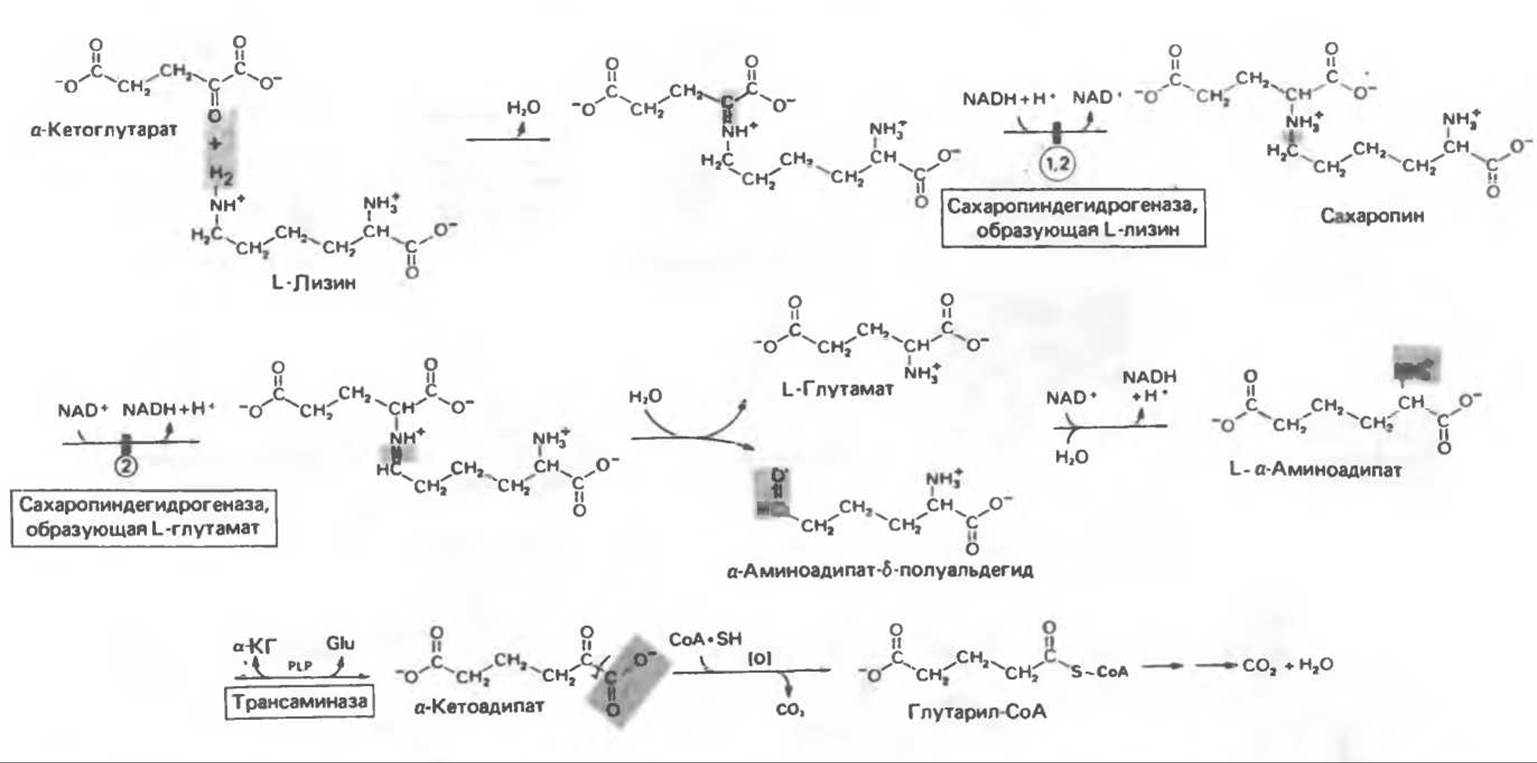
Рис. 31.18. Катаболизм L-лизина (а-КГ — а-кетоглутарат, Glu — глутамат, PLP — пиридоксальфосфат). Цифры в кружочках указывают вероятные места метаболических нарушений при 1 — периодической гиперлизинемии с сопутствующей гипераммонемией, 2 — устойчивой гиперлизинемии без сопутствующей гипераммонемии.
А. Периодическая гиперлизинемия и сопутствующая гипераммониемия. При периодической гиперлизинемии даже потребление белка в нормальных количествах вызывает повышение концентрации лизина в тканях белка. Вторичной по отношению к гиперлизинемии является гипераммониемия, развивающаяся вследствие конкурентного ингибирования лизином активности печеночной аргиназы (см. рис. 30.13).
Увеличение потребления жидкости и снижение содержания лизина в диете понижают уровень гипераммониемии и ослабляют ее клинические проявления. И наоборот, нагрузка лизином вызывает тяжелый криз и коматозное состояние. Пока не имеется данных о генетической природе болезни.
Б. Стойкая гиперлизинемия без сопутствующей гипераммониемии. Клинические и биохимические данные оказались весьма варьирующими у 12 пациентов со стойкой гиперлизинемией. У некоторых больных, но не у всех, наблюдалась умственная отсталость, Сопутствующей гипераммониемии не отмечалось даже после лизиновой нагрузки. В ряде случаев наблюдалось накопление катаболитов лизина в биологических жидкостях. Устойчивую гиперлизинемию рассматривают как аутосомно-рецессивный признак. Наряду с блокированием процесса превращения лизина и а-кетоглутарата в сахаропин у некоторых пациентов, по-видимому, имелось нарушение превращения сахаропина в L-глутамат и а-аминоадипат-8-полуальдегид (рис. 31.18).
Триптофан
Триптофан вследствие многообразия связанных с ним метаболических реакции и продуктов был одной из первых аминокислот, которые были отнесены к незаменимым. Исследования, проведенные на мутантах Neurospora и на бактериях Pseudomonas, а также выделение метаболитов триптофана из мочи, оказали неоценимую помощь в выяснении деталей метаболизма триптофана.
При введении с пищей [14С]-триптофана большая часть изотопа включается в состав белков, однако существенная часть обнаруживается в моче в составе различных катаболитов. Атомы углерода боковой цепи и ароматического кольца могут полностью переходить в амфиболические интермедиаты при трансформации триптофана по кинуренин-антранилатному пути (рис. 31.19), играющему важную роль в деградации триптофана и в его превращении в никотинамид.
Триптофаноксигеназа (триптофанпирролаза) катализирует раскрытие индольного кольца с включением двух атомов молекулярного кислорода в образующийся N-формилкинуренин. Данный фермент является металлопротеином, содержащим железо-порфирик; синтез его в печени индуцируется адрено-кортикостероидами и триптофаном. Значительная часть синтезированного фермента находится в латентной форме и требует активации. Триптофан стабилизирует оксигеназу по отношению к протеолитическим ферментам. Она ингибируется по принципу обратной связи производными никотиновой кислоты, в том числе NADPH.
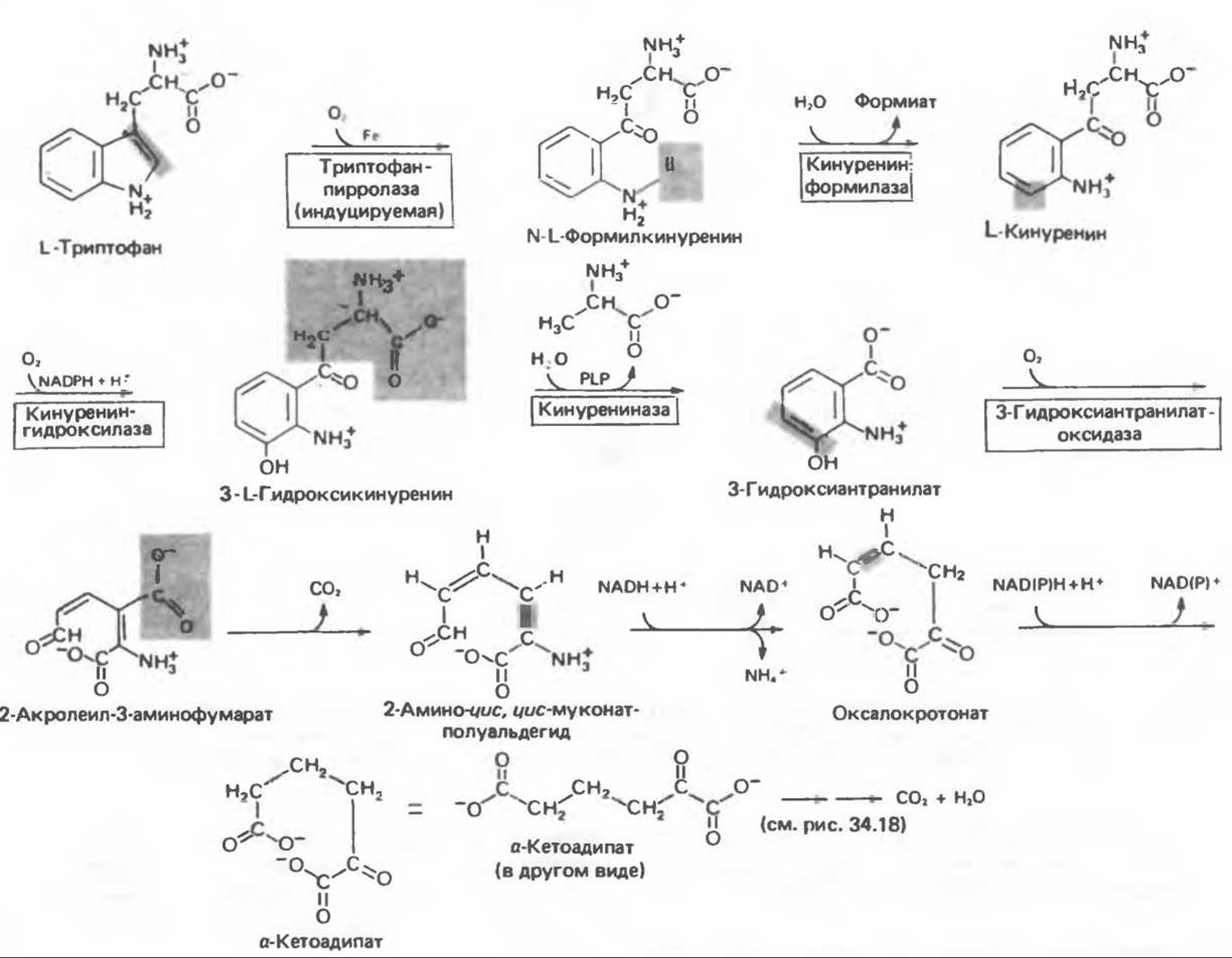
Рис. 31.19. Катаболизм L-триптофана PLP-пиридоксальфосфат.
Гидролитическое удаление формильной группы N-формилкинуренина катализируется в печени млекопитающих кинуренин-формилазой. Гидролиз в Н218О приводит к включению атома 18О в образующийся формиат. Фермент также катализирует аналогичные реакции с различными арилформиламинами.
Продуктом реакции, катализируемой кинуренин-формилазой, является кинуренин (рис. 31.19). Он может быть дезаминирован в результате реакции переаминирования с переносом аминогруппы боковой цепи на а-кетоглутарат. Образующееся при этом кетопроизводное претерпевает спонтанную циклизацию, превращаясь в кинуреновую кислоту. Это соединение является побочным продуктом катаболизма кинуренинт и не относится к катаболитам, образующимся на главном пути (рис. 31.19).
Дальнейший ход метаболизма кинуренина включает его превращение в 3-гидроксикинуренин и далее в 3-гидроксиантранилат. Гидроксилирование происходит при участии молекулярного кислорода и осуществляется в NADPH-зависимой реакции гидроксилирования, аналогичной реакции гидроксилирования фенилаланина (см. гл. 29).
Кинуренин и гидроксикинуренин превращаются в гидроксиантранилат при участии пиридоксальфосфат-содержащего фермента кинурениназы. Недостаток витамина В6 приводит к частичной утрате способности к катаболизму этих кинурениновых производных; во внепеченочных тканях они превращаются в ксантуренат (рис. 31.20). Этот в норме отсутствующий катаболит появляется в моче человека обезьян и крыс при недостаточном содержании в пище витамина В6. В этих условиях введение избыточных количеств триптофана приводит к экскреции ксантурената с мочой.

Рис. 31.20. Образование ксантурената в условиях недостаточности витамина В6. Нарушено превращение катаболита триптофана 3-гидроксикинуренина в 3-гидроксиантранилат (рис. 31.19). Поэтому большая часть катаболита превращается в ксантуренат.
У многих животных превращение триптофана в никотиновую кислоту делает необязательным поступление этого витамина с пищей. У крыс, кроликов, собак и свиней пищевой триптофан может полностью заменить этот витамин; у человека, а также у ряда животных избыточное потребление триптофана с пищей повышает экскрецию с мочой производных никотиновой кислоты (например, N-металникотинамида). При недостатке витамина В6 нарушение образования из триптофана никотиновой кислоты может привести к нарушению синтеза пиридиновых нуклеотидов, NAD+ и NADP+. Если ввести в организм достаточное количество никотиновой кислоты, нормальный синтез пиридиновых нуклеотидов возобновляется даже в отсутствие витамина В6.
Метаболические нарушения катаболизма триптофана. Болезнь Хартнупа, наследственное нарушение метаболизма триптофана, характеризуется появлением сыпи на коже, как при пеллагре, перемежающейся мозжечковой атаксией и умственной отсталостью. Моча больных содержит значительно повышенные количества индолацетата (а-N-[индол-3-ацетил]глутамина) и триптофана.