Биохимия человека Том 2 - Марри Р. 1993
Структура, функция и репликация информационных макромолекул
Технология рекомбинантных ДНК
Генная инженерия и анализ молекулярной природы заболеваний
Нормальные генетические варианты
Существуют нормальные варианты последовательностей ДНК человека (полиморфизм). Такие варианты встречаются примерно 1 раз на 500 нуклеотидов или 107 раз на геном. Сюда входят делеции, вставки, а также единичные замены нуклеотидов. У здоровых людей эти изменения либо не затрагивают кодирующих последовательностей, либо происходят в функционально несущественных участках кодирующих областей ДНК. Полиморфизм структуры ДНК может быть прямо связан и с определенными заболеваниями. В последние годы феномен полиморфизма все чаще используется для идентификации соответствующих специфических генов.
Генетические изменения, вызывающие заболевания
На более ранних этапах развития медицинской генетики существовало представление, что большинство наследственных болезней вызвано толковыми мутациями, которые проявляются в функциональном несовершенстве соответствующего измененного белка. Роль толковых мутаций в возникновении наследственных патологий действительно велика, к этому следует добавить только, что генетические заболевания могут быть вызваны нарушениями на любой стадии процесса, представленного на рис. 36.1. Это положение хорошо иллюстрируется исследованиями гена ß-глобина, имеющего кластерную организацию и картированного на одиннадцатой хромосоме (рис. 36.7, 36.8). Биосинтез дефектного ß-глобина служит причиной целого ряда заболеваний. Патологические симптомы могут быть связаны с нарушениями как в самом гене, так и в его окружении (табл. 36.6).
Точковые мутации
Классический пример заболевания, связанного с толковой мутацией, — серповидноклеточная анемия. Болезнь вызывается заменой всего лишь одного из 3х109 оснований, составляющих полный геном человека: в шестом кодоне ß-глобинового гена аденин заменяется на тимин. С измененного кодона считывается не глутаминовая кислота, а валин, что приводит к структурным нарушениям молекулы ß-глобина. Некоторые толковые мутации вызывают снижение либо полную остановку синтеза ß-глобина. Результатом таких нарушений является ß-талассемия (талассемии — класс заболеваний, связанных с нарушениями синтеза глобина). На рис. 36.8 представлены позиции толковых мутаций, нарушающих какую-либо из многочисленных стадий процесса образования нормальной ß-глобиновой мРНК и, следовательно, вызывающих ß-талассемию.

Рис. 36.8. Мутации гена ß-глобина, обусловливающие ß-талассемию. Ген представлен в ориентации 5' → 3'. Заштрихованы нетранслируемые 5'- и 3'-области. При чтении в направлении 5' → 3' затемненные участки — экзоны 1—3, светлые участки интроны 1 и 2. Мутации, затрагивающие контроль транскрипции (●), локализованы в 5'-фланкирующей области. Идентифицированы отмеченные на рисунке некоторые nonsense-мутации (∆), мутации, влияющие на процессинг (◊) и расщепление РНК (О). В некоторых областях выявлено большое количество мутаций. Такие участки помечены квадратными скобками.
Таблица 36.6. Структурные изменения в гене ß-глобина
|
Изменение |
Затронутая функция |
Заболевание |
|
Точковые мутации |
Сворачивание белковой глобулы |
Серповидноклеточная анемия |
|
Контроль транскрипции |
ß-Талассемия |
|
|
Сдвиг рамки и мутации |
ß-Талассемия |
|
|
Депеция |
Образование мРНК |
ß0-Taлacceмия |
|
Гемоглобин Лепоре |
||
|
Перестройка |
Образование мРНК |
ß-Талассемия тип III |
Делеции, вставки и перестройки ДНК
Исследования геномов бактерий, вирусов, дрожжей и дрозофилы показывают, что отдельные участки ДНК могут менять свое положение в геноме. Утрата функционально важного участка ДНК, перестановка фрагментов ДНК внутри гена, вставка в его кодирующий или регуляторный участок, как правило, вызывают изменения в уровне экспрессии данного гена, приводящие к заболеванию. Молекулярный анализ ß-талассемии выявляет большое число подобных случаев (особенно делеций). Создается впечатление, что кластеры глобиновых генов весьма подвержены повреждениям. Делеции в аглобиновом кластере, локализованном в 16-й хромосоме, приводят к а-талассемии. Для большинства таких делеций отмечена четкая взаимосвязь с этническим происхождением. Жители севера Европы, филиппинцы, негроиды и представители средиземноморских популяций имеют нарушения различного типа, но все они ведут к утрате гемоглобина А и а-талассемии.
Подобный анализ можно провести и для многих других заболеваний. Обычно точковые мутации идентифицируют путем определения нуклеотидной последовательности изучаемого гена, но, если мутация затрагивает сайт рестрикции, для ее выявления достаточно рестрикционного анализа. Делеции и вставки ДНК-фрагментов больших чем 50 пар оснований определяют методом блоттинга по Саузерну.
Анализ родословных
На примере серповидноклеточной анемии можно убедиться в том, насколько эффективен генноинженерный подход при изучении болезней человека. Замена основания в кодирующей цепи ДНК гена гемоглобина приводит к изменению последовательности, соответствующей шестому кодону:

При этом исчезает сайт рестриктазы Mst 11 (ССТ- NAGG; стрелками указаны места расщепления, см. табл. 36.1). Другие Mst Н-сайты (рис. 36.9) остаются неизменными и могут быть расщеплены этой рестриктазой. Поэтому Саузерн-блоттинг обработанных ферментом образцов ДНК нормальных (АА), гетерозиготных (AS) и гомозиготных (SS) пациентов дает три типа распределения фрагментов (рис. 36.9). Этот пример показывает, как на уровне ДНК можно провести анализ родословной с использованием описываемых в этой главе принципов и подходов. Такой анализ весьма эффективен при изучении ряда генетических заболеваний, вызванных делениями, вставками и (реже) точковыми мутациями с изменением сайтов рестрикции (как в только что рассмотренном примере).
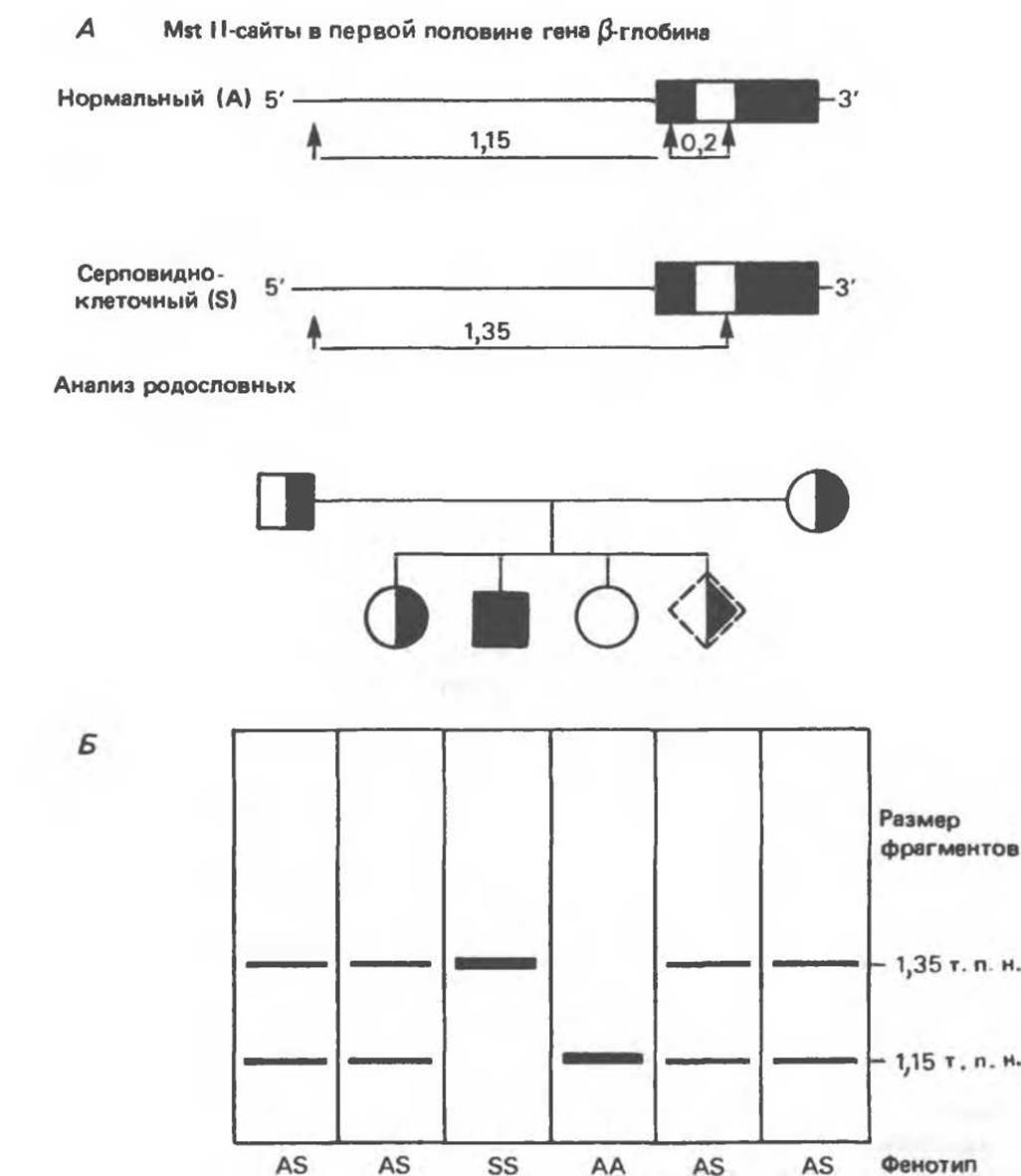
Рис. 36.9. Анализ родословных в случае серповидноклеточной анемии. В верхней части рисунка (А) показано начало гена ß-глобина с сайтами расщепления рестриктазой Mst II (Т) У нормального (А) и серповидноклеточного (S) ß-глобина. В результате расщепления ДНК здоровых индивидуумов рестриктазой Mst II образуются специфические фрагменты ДНК размером 1,15 и 0.2 т. п. н. Замена одного основания у больных серповидноклеточной анемией приводит к потере одного из трех Mst II-сайтов в области гена и соответственно к появлению только одного специфического Mst II-фрагмента размером 1,35 т. п. н. Это различие в длине легко обнаруживается методом Саузерн-блоттинга (Б). (На данном рисунке положение фрагмента длиной 0,2 т. п. н. не указано.) Анализ родословных демонстрирует три возможных генотипа АА-норма (О), AS-гетерозигота по гену серповидноклеточности (◑◨) и SS-гомозигота по гену серповидных эритроцитов (■). Этот подход позволяет осуществлять пренатальную диагностику заболевания серповидноклеточной анемией и выявлять гетерозиготных носителей соответствующего гена (◆).
Пренатальная диагностика
Дородовая диагностика наследственных заболеваний возможна, если известна природа генетического нарушения и имеется соответствующий зонд. Анализу по Саузерну можно подвергнуть ДНК клеток, собранных из 10 мл амниотической жидкости (или полученных с помощью биопсии ворсинок хориона). Плод с рестрикционным вариантом АА (рис. 36.9) нормален и не является носителем серповидноклеточной анемии. В случае варианта SS можно с уверенностью предсказать развитие болезни. Уже сейчас существуют зонды для подобного анализа многих заболеваний.
Полиморфизм длины рестрикционных фрагментов (ПДРФ)
Изменения в последовательностях ДНК, вызванные описанными выше причинами, могут обусловливать изменения в расположении сайтов рестрикции и поэтому сказываться на длине рестрикционных фрагментов. Устойчивое наследуемое изменение в распределении длин рестрикционных фрагментов (наблюдаемое для более чем 1 % численности популяции) называют полиморфизмом длины рестрикционных фрагментов (ПДРФ). Этот феномен может быть результатом либо толковых замен (серповидноклеточная анемия), либо делеций и вставок (талассемии). В последнее время ПДРФ стали с успехом использовать в диагностических целях. Рестрикционный полиморфизм обнаружен как в последовательностях известных генов, так и в участках ДНК с неизвестной функцией. ПДРФ может нарушать биологическую функцию, а может и не иметь никаких биологических последствий; в любом случае соответствующие измененные локусы наследуются в соответствии с законами Менделя. Основная область использования этого феномена (а известно уже около 350 случаев ПДРФ) — диагностика наследственных заболеваний, функциональная природа которых неизвестна. Вначале с помощью ПДРФ устанавливают группу сцепления, а затем методом последовательной гибридизации определяют локус, ответственный за заболевание. Согласно методу последовательной гибридизации, фрагмент, представляющий один из концов длинной цепи ДНК, используют в качестве зонда для выявления другого фрагмента, который перекрывается с первым и в то же время выходит за его пределы. Применение этого подхода, называемого «прогулка по хромосоме» (рис. 36.10), обеспечивает «передвижение» по цепи ДНК вплоть до интересующей области. Направление передвижения контролируется по рестрикционной карте. «Прогулка по хромосоме» особенно удобна при изучении Х-сцепленных болезней, поскольку в данном случае экспрессируется только один из двух аллелей. 20% выявленных случаев ПДРФ относится именно к Х-хромосоме, и именно для нее составлена практически полная карта. Используя феномен ПДРФ, можно локализовать ген любой X-сцепленной болезни (например, мышечной дистрофии Дюшенна). С помощью ПДРФ удалось установить, что генетический дефект при хорее Гентингтона затрагивает конец короткого плеча хромосомы 4, а ген, вызывающий поликистоз почек, сцеплен с локусом а-глобина на хромосоме 16.
Генная терапия
Заболевания, вызванные функциональной недостаточностью продукта того или иного гена, можно лечить с помощью «заместительной» терапии (табл. 36.5). Стратегия подхода заключается в клонировании гена (например, гена, кодирующего аденозиндезаминазу) в векторе, способном включиться в геном клетки хозяина. Весьма перспективным представляется использование для этого предшественников клеток костного мозга. Можно надеяться, что такие клетки «приживутся» и будут размножаться, синтезируя трансгенный продукт. Конечно, ген, перенесенный в соматические клетки, потомкам не передается.
В последнее время ведутся интенсивные поиски путей генно-инженерного воздействия на половые клетки. Соответствующие эксперименты проводят на лабораторных животных. Гены, инъецированные в оплодотворенные яйцеклетки мыши, в некоторых случаях встраиваются в геном. Полученных трансгенных животных используют для изучения характера экспрессии генов в разных тканях, а также для выявления специфических генов онтогенеза. Трансгенный подход недавно с успехом был использован для коррекции генетического дефекта у мышей. В оплодотворенные яйцеклетки мыши с наследственным гипогонадизмом (гл. 55) инъецировали ДНК, содержащую кодирующую последовательность предшественника гонадолиберина. У части развившихся из таких яйцеклеток мышей этот ген нормально экспрессировался. Фенотипически эти мыши были нормальными во всех отношениях. Их потомство также не проявляло недостаточности по гонадолиберину. Приведенный пример свидетельствует о принципиальной возможности экспрессии трансгенов в соматических клетках и их передачи потомству.
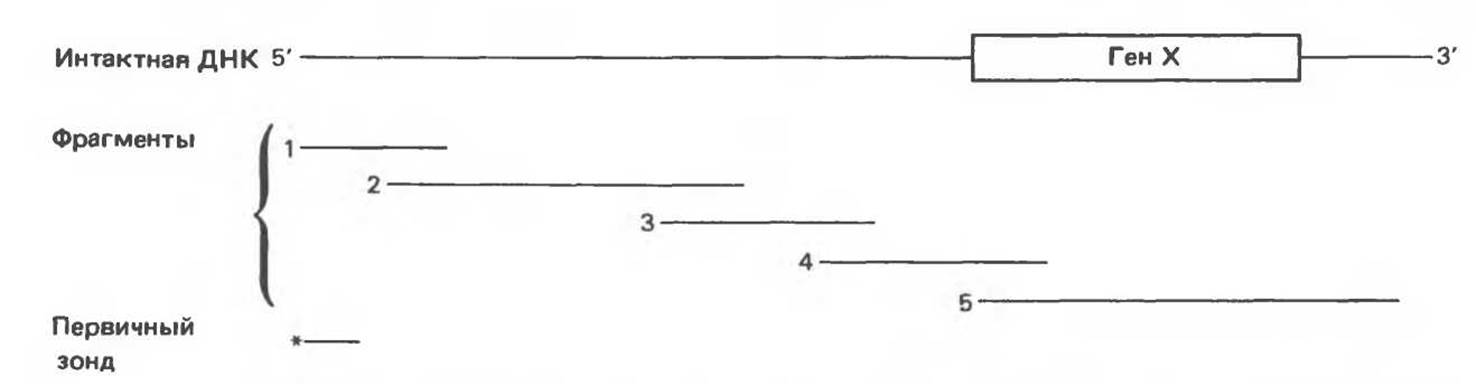
Рис. 36.10. Метод «прогулка по хромосоме». Пусть необходимо обнаружить ген X в рамках протяженного фрагмента ДНК. Точное положение гена неизвестно, однако имеется первичный зонд (*), соответствующий некоему участку генома (показан в данном случае на 5'-конце исследуемого фрагмента ДНК). Кроме того, имеется библиотека перекрывающихся фрагментов генома. (Для упрощения на рисунке изображены только пять фрагментов.) Первичный зонд гибридизуется только с клонами, содержащими фрагмент 1. Этот фрагмент можно использовать далее в качестве зонда для выявления фрагмента 2. Процедура последовательной гибридизации повторяется вплоть до обнаружения фрагмента 4, который гибридизуется с фрагментом 5, содержащим искомый ген X.