Биохимия - Химические реакции в живой клетке Том 2 - Д. Мецлер 1980
Биосинтез; как образуются новые молекулы
Внутриклеточное расщепление полисахаридов и гликолипидов
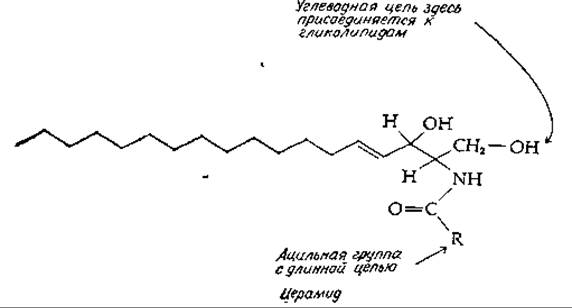
Гликолипиды
Предполагается, что гликолипиды, подобно гликопротеидам, синтезируются на мембранах эндоплазматического ретикулума, причем образовавшиеся гликолипиды размещаются на поверхности мембраны, выстилающей цистерны эндоплазматического ретикулума. Отсюда они транспортируются в аппарат Гольджи и в конце концов выходят наружу, включаясь в состав наружной поверхности плазматической мембраны. Основной класс гликолипидов — это сфинголипиды, представленные цереброзидами и ганглиозидами. И цереброзиды, и ганглиозиды образуются из церамидов — производных N-ацилированных сфингозинов (табл. 2-8). На рис. 12-5 изображены некоторые пути биосинтеза этих соединений из сфингозина. Источниками ацильных, гликозильных и сульфатных групп служат соответствующие производные СоА, CDP, UDP, СМР. На этой схеме показан также биосинтез сфингомиелина, а также З-фосфоаденозин-5-фосфосульфат, который мы рассмотрим в этой главе.
Каждый этап биосинтеза, изображенного на рис. 12-5, требует специфической трансферазы. Известно, что большая часть трансфераз связана с мембранами, но подробные сведения об этих ферментах отсутствуют. Кроме того, вполне вероятно, что последовательность действия трансфераз не всегда строго фиксирована1), а вся схема биосинтеза может быть значительно сложнее, чем это показано на рисунке.
Удивительный аспект метаболизма гликолипидов заключается в существовании по крайней мере десяти специфических лизосомных болезней накопления — сфинголипидозов [24—28]. Биохимические нарушения, лежащие в основе этих заболеваний, указаны на рис. 12-5 и в табл. 12-1. Первым изученным сфинголипидозом была болезнь Гоше, наследуемая по аутосомному рецессивному типу. При этой болезни происходит избыточное накопление глюкозилцерамида, приводящее в конечном итоге к значительному поражению печени и селезенки, причем последняя у людей, заболевших в зрелом возрасте, увеличивается в 4—5 раз по сравнению с нормой. У детей болезнь протекает в более тяжелой форме, чем у взрослых, и сопровождается задержкой умственного развития. Как показал Брэди (Brady), скорость синтеза цереброзидов у этих больных остается на уровне нормы. В дальнейшем (в 1965 г.) удалось выяснить, что при болезни Гоше отсутствует одна из лизосомных гидролаз, участвующая в катаболизме гликолипидов (блок № 3 на рис. 12-5, где реакции катаболизма показаны пунктирными стрелками); в итоге путь их распада оказывается блокированным.
1) Примером другой последовательности биосинтетических реакций может служить образование галактозилцерамида путем переноса галактозы на сфингозин с последующим ацилированием:

Однако процесс, изображенный на рис. 12-5, имеет, по-видимому, более важное значение.
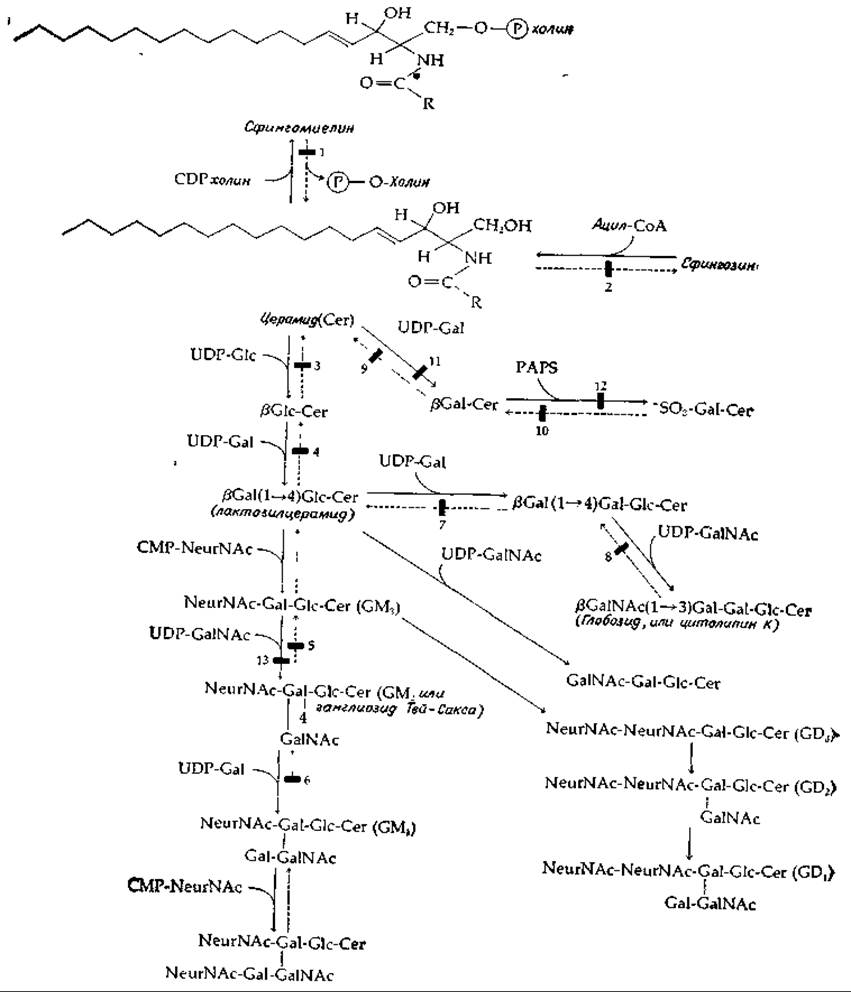
РИС. 12-5. Биосинтез и распад гликосфинголипидов. Жирной чертой показаны участки, блокированные при болезних, обусловленных нарушениями обмена веществ.
Таблица 12-1 Болезни накоплении гликолипидов (сфинголипидозы)
|
Номер на рис. 12-5 |
Наименование |
Поврежденный фермент |
|
1. Болезнь Нимана — Пика |
Сфингомнелиназа |
|
|
2. Болезнь Фабера (липогрануломатоз) |
Церамидазаа |
|
|
3. Болезнь Гоше |
ß-Глюкозидаза |
|
|
4. Галактознлцерамидоз |
ß-Галактозилгидролаза |
|
|
5. Болезнь Тей — Сакса |
Гексозаминидаза Аб |
|
|
6. Генерализованный ганглиозидоз |
ß-Галaктозидазав |
|
|
7. Болезнь Фабри |
а-Галактозидазаг,в |
|
|
8. Болезнь Сандхофа |
Гексозаминидазы А и В |
|
|
9. Лейкодистрофия Краббе |
Галактоцереброзидаза |
|
|
10. Метахроматическая лейкодистрофия |
Сульфатаза |
|
|
13. Накопление гематозида (GM3) |
GМ3-N-ацетилгалактозаминил — трансферазае |
|
а Sugita М., Dulaney І. Г., Moser ff. W., Science, 178, 1100—1102 (1972).
б Tollman F., Brady R. O., JBC, 247, 7570—7575 (1972).
в Этот же дефект обнаружен у одной из линий сиамских кошек; Baker Н. J., Jr., Lindsey J. R., McKhann О. М., Farrell D. F., Science, 174, 838—839 (1971).
г Beutler Е.. Kuhl V., JBC, 247, 7195—7200 (1972).
д Crawhall C., Banfalvt M., Science, 177,527—528 (1972).
e Fishman P. H., Max S. R., Tollman І. F., Brady R О, Maclaren N. K., Comblath M, Science, 487, 68—70 (1975).
Известна также болезнь Фабри, в основе которой лежит дефект сцепленного с Х-хромосомой гена, обеспечивающего отщепление галактозильных остатков от цереброзидов. В результате накапливается тригликозилцерамид, распад которого блокирован на уровне реакции 7 (рис. 12-5).
Чаще всего встречающимся и наиболее изученным сфинголипидозом является болезнь Тей — Сакса. Впервые она была описана в 1881 г., и с тех пор было описано более 500 случаев этого заболевания. Это тяжелая болезнь, сопровождающаяся задержкой развития головного мозга, слепотой, параличом, слабоумием; больные погибают в возрасте около 3 лет. Ежегодно в Северной Америке рождается примерно 30 детей с этой патологией, а в целом на земном шаре эта цифра, вероятно, в 5—7 раз больше.