Биохимия - Химические реакции в живой клетке Том 2 - Д. Мецлер 1980
Ферменты: белковые катализаторы клеток
Механизмы ферментативного катализа
Кислотный и основный катализ
Многие реакции, которые ускоряются ферментами, могут катализироваться также кислотами или основаниями, а часто и соединениями обоих типов. Хорошо изученным примером такого рода является мутаротация — обратимое взаимное превращение а- и ß-аномерных форм сахаров, в частности глюкозы [см. схему (6-75)]. Эта реакция катализируется специфическим ферментом мутаротазой, а также неорганическими кислотами и основаниями. Эти данные показывают, что между простыми кислотами и основаниями, с одной стороны, и ферментами — с другой, есть нечто общее с точки зрения каталитического действия. Поскольку многие боковые цепи аминокислот содержат кислотные и основные группы, мы приходим к вполне естественному заключению, что эти группы должны участвовать в катализе как кислоты и основания. Однако для того чтобы понять, как именно они участвуют в катализе, мы должны иметь представление о численных значениях некоторых констант равновесия и констант скорости.
а. Сильные и слабые кислоты
Кислоты являются донорами протонов, а основания — их акцепторами. Кислота может быть охарактеризована константой диссоциации или взятым с противоположным знаком десятичным логарифмом этой константы рКа (см. также гл.4, разд В):
![]()
где Ка = [Н+] [В-]/[НВ] Заметим, что [Н+] представляет собой концентрацию (или, точнее говоря, активность) ионов Н3О+, а не свободных протонов. Далее мы будем обозначать кислоты символами НВ или Н+В, а сопряженные основания, образующиеся при диссоциации кислот, — символами В- или В.
Напомним, что сильные кислоты имеют низкие значения рКа, а сопряженные основания являются слабыми основаниями. В то же время очень слабые кислоты имеют высокие значения рКа, а сопряженные основания являются сильными основаниями. При рассмотрении механизмов ферментативных реакций целесообразно знать значения рКа следующих групп:

Эти значения не являются постоянными и могут быть больше или меньше на 0,5 единицы в зависимости от структуры соединения и окружения данной боковой группы в белке. В некоторых случаях отклонения бывают даже более существенными.
б. Кислотно-основный катализ мутаротации
Процесс мутаротации глюкозы протекает через стадию образования в качестве промежуточного соединения свободной альдегидной формы:

В ходе реакции от ОН-группы при С-1 атоме а-глюкозы отрывается водородный атом и протон (по-видимому, иной) переносится на кислодор кольца с разрывом С—О-связи. Обратный процесс может проходить также по пути б, что ведет к образованию ß-изомера.
Перенос протона (чаще всего между атомами кислорода, азота и серы) происходит в ходе многих биохимических реакций. Связи между атомом водорода, с одной стороны, и атомами кислорода, азота и серы — с другой, обычно сильно поляризованы, что приводит к появлению на водородных атомах небольшого положительного заряда. Таким образом, группы приобретают слабо кислотный характер, и протоны могут сравнительно легко отрываться от них и переноситься на другие группы. Естественно допустить, что каталитические свойства кислот и оснований связаны с подобным переносом водородных атомов.
в. Общий основный и общий кислотный катализ
Катализируемая основанием мутаротация протекает следующим образом: основание, например ОН--ион, атакует протон гидроксильной группы; в результате последняя превращается в анион и одновременно образуется сопряженная кислота ВН+ (стадия а на приведенной ниже схеме):
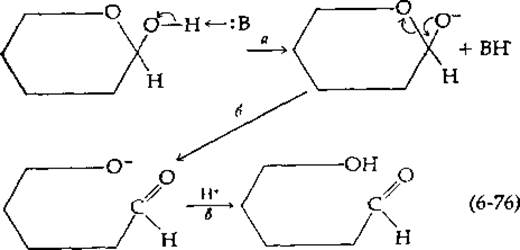
Анион затем изомеризуется в форму с раскрытым кольцом (стадия б). Присоединение протона (в результате переноса от Н3О+) приводит к образованию свободной альдегидной формы сахара (стадия в).
В роли основания-катализатора в этой схеме может выступать не Только ОН--ион, но и другие, более слабые основания, например ион аммония или даже вода. Как оказалось, в некоторых случаях скорость каталитической реакции пропорциональна только концентрации ионов ОН-, а присутствие других, более слабых оснований на этой величине не сказывается. Подобный катализ называют специфическим катализом гидроксильным ионом. В общем случае кажущаяся константа скорости реакции первого порядка (kobs) для изучаемого процесса представляет собой сумму нескольких членов:
kobs = kH2O + kOH-[OH-] + kB[В]. (6-77)
Член kH2O характеризует скорость процесса в чистой воде и соответствует каталитическому действию одной воды либо как кислоты, либо как основания. Два последних члена определяют вклад в катализ соответственно ОН--иона и другого основания. Член kB[B] характеризует общий основный катализ, который, как полагают, играет важную роль в функционировании многих ферментов. В энзимологии под общим основным катализом подразумевают способность некоторой основной группы в молекуле фермента выполнять роль акцептора протона.
Катализ мутаротации кислотами наблюдается в том случае, когда кислота служит донором протона, присоединяющегося к атoму кислорода в кольце молекулы сахара:
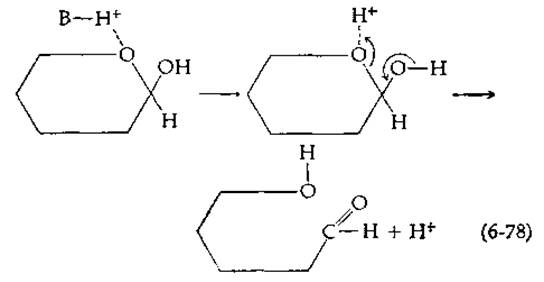
Как и в предыдущем случае, возможен либо специфический кислотный катализ (под действием иона Н3O+), либо общий кислотный катализ.
Можно полагать, что при функционировании ферментов реализуются общий основный и общий кислотный катализ. Ферменты не способны локально концентрировать протоны или гидроксильные ионы с тем, чтобы обеспечить специфический основный или специфический кислотный катализ. Однако определенные ионогенные группы ферментов при их нормальной степени протонирования, соответствующей pH клетки, могут выступать в роли общекислотных и общеосновных катализаторов.
г. Согласованные механизмы
Третий возможный тип катализа предполагает синхронное действие основания и кислоты, обусловливающее одновременный разрыв старой связи и образование новой. Известно, например, что мутаротация тетраметилглюкозы в бензоле, содержащем либо пиридин (основание), либо фенол (кислоту), протекает очень медленно. Однако, когда в растворе одновременно присутствуют и пиридин, и бензол, мутаротация протекает существенно быстрее. На основании этого факта Свейн и Браун [50] предложили согласованный механизм, в котором одновременно участвуют и кислота, и основание:
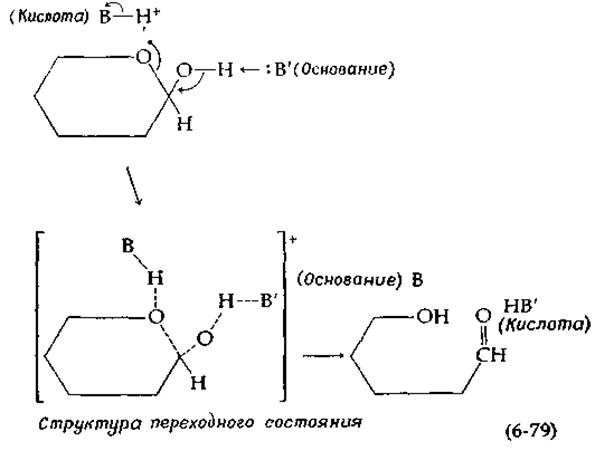
Заметим, что в ходе реакции кислота ВН+ превращается в сопряженное основание В, а основание В' — в сопряженную кислоту НВ'. Может показаться, что, поскольку эти агенты изменяются в ходе реакции, они не являются истинными катализаторами. Однако простой обмен протонов восстанавливает исходные формы и завершает каталитический цикл. В водных растворах вода сама может выступать в роли кислоты или основания или даже одновременно в роли кислоты и основания при согласованном катализе.
Экспериментальные данные о наличии согласованного кислотно-основного катализа процесса мутаротации тетраметилглюкозы в бензоле фенолом и пиридином считаются в настоящее время недостаточно убедительными [51, 52]. Для неферментативных реакций, протекающих в водных растворах, доказать существование такого механизма катализа весьма трудно1). Однако он может играть исключительно большую роль в случае ферментативного катализа, поскольку среди боковых групп аминокислот могут найтись две соответствующим образом расположенные кислотные и основные группы.
д. Соотношения Брёнстеда
Эффективность данного основания как общего основного катализатора можно обычно связать с его основностью (рКа), воспользовавшись уравнением Брёнстеда [53]:
lgkB = lgGB + ß(pKa). (6-80
Константа kB в этом уравнении определяется выражением (6-77), а GB представляет собой постоянную величину для реакции данного типа. Аналогичное соотношение связывает константу скорости kA для общего кислотного катализа с величиной рKа:
lg kHB = lg GA — а (рКа). (6-81)
Эти уравнения подобны линейным соотношениям, в которые входит свободная энергия (гл. 3, разд. Д), однако они касаются скоростей, а не равновесий. Правда, уравнение Гаммета [уравнение (3-66)] также очень часто применяется не только к константам равновесия, но и к константам скорости.
Условием выполнимости уравнений Брёнстеда является существование прямой связи между свободной энергией активации реакции и основностью (или кислотностью) катализатора.
Коэффициенты β и а в уравнениях (6-80) и (6-81) характеризуют чувствительность скорости реакции к изменению основности или кислотности катализатора. Нетрудно показать, что в том случае, когда ß или а близки к 1, общий основный или общий кислотный катализ обычно отсутствуют и скорость реакции определяется только специфическим катализом гидроксильным или водородным ионом [53]. При уменьшении ß или а до 0 вклад основного или кислотного катализа тоже становится исчезающе малым. Таким образом, общий основный или общий кислотный катализ наиболее существен при значениях коэффициентов ß и а, близких к 0,5. При этих условиях, как нетрудно видеть, такое относительно слабое основание, как имидазол (в боковой цепи гистидина), может оказаться необычайно эффективным катализатором при pH 7.
1) Недавно, правда, было показано, что катализ енолизации ацетона уксусной кислотой и ацетатом действительно протекает по согласованному кислотно-основному пути [52а].
Для экспериментального определения значений ß и а необходимо построить график зависимости lgkBили lgkHBот рKа (график Брёнстеда) и определить наклон полученной прямой. В случае ионизированных аминогрупп, в которых от атома азота может отщепиться один из трех протонов, и дикарбоновых кислот нужно вводить статистические поправки (гл. 4, разд. В, 5).
Любой механизм, для которого выполняется общий основный или общий кислотный катализ, характеризуется одним важным свойством: отщепление или присоединение протона происходит на стадии, лимитирующей скорость реакции.
е. Таутомерный катализ
Свейн и Браун [50] провели весьма интересный эксперимент, показавший, что кислотная и основная группы, включенные в одну и ту же молекулу, катализируют мутаротацию сахаров гораздо эффективнее, чем простая смесь кислоты и основания. Так, 0,001 М а-оксипиридин катализирует мутаротацию тетраметилглюкозы (0,1 М) в бензоле в 7000 раз более эффективно, чем смесь, содержащая 0,001 М пиридин и 0,001 М фенол. Свейн и Браун предложили следующий полностью согласованный механизм реакции для полифункционального катализатора а-оксипиридина. Они допустили, что реакции предшествует образование стабилизированного водородными связями комплекса, аналогичного фермент-субстратному комплексу:
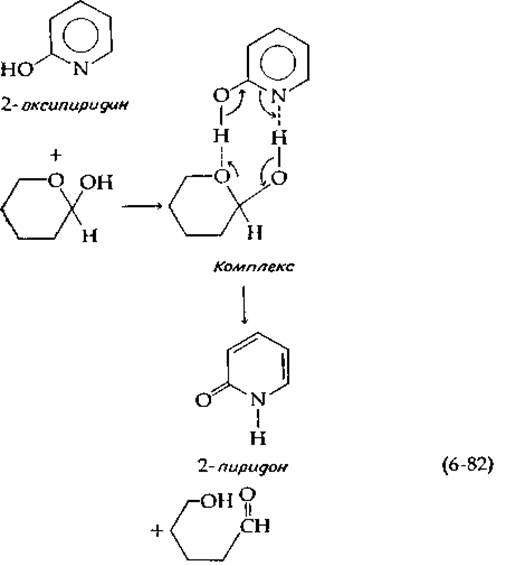
продуктом превращения катализатора является 2-пиридон — таутомерная форма 2-оксипиридина, причем между обеими формами быстро устанавливается равновесие.
Рони [51] предложил называть катализ а-оксипиридином таутомерным катализом; он считает, что высокая эффективность этого катализатора обусловлена не просто близостью кислотной и основной групп в одной и той же молекуле, но и способностью катализатора совершать обратимые переходы из одной таутомерной формы в другую.
Предельное значение для констант скорости второго порядка в условиях, когда скорость реакции лимитируется диффузией, составляет приблизительно 1010 М-1∙с-1 (разд. А, 7). В 1956 г. Эйген [54], разработавший новые методы изучения быстрых реакций, сделал удивительное открытие, обнаружив, что протоны и гидроксильные ионы взаимодействуют гораздо быстрее, находять в решетке льда, чем в растворе: константы скорости второго порядка составляли 1013—1014 М-1∙с-1. Соответствующие им скорости реакции так же высоки, как скорости молекулярных колебаний; например, частота колебаний ОН-связи в воде2) составляет примерно 1014 с-1. Объясняется этот факт, по-видимому, следующим. ОН--ион и протон, который присоединяется к молекуле воды с образованием иона Н3O+, связаны водородными связями с соседними молекулами воды. Поскольку во льду водородными связями соединены все молекулы воды, гидроксильные и водородные ионы оказываются соединенными цепочкой из молекул воды:
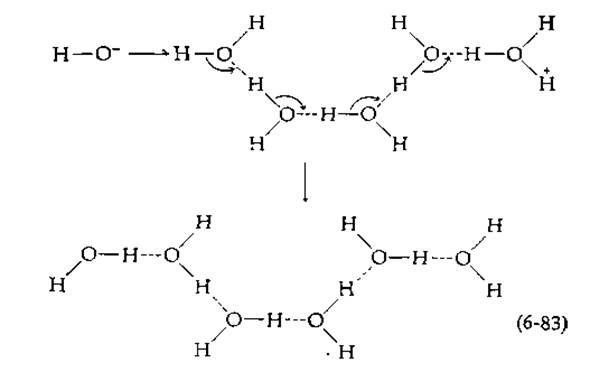
ж. Скорости переноса протона1)
Благодаря синхронному перемещению электронов от ОН--иона через цепочку (маленькие стрелки на схеме) во время одного колебания может произойти нейтрализация зарядов. Заметим, что положения атомов кислорода к концу процесса остаются прежними, в то время как протоны, вовлеченные в образование водородных связей, слегка перемещаются на схеме влево. Реакция подобного типа не только является свидетельством удивительной подвижности водородных ионов, но и может иметь прямое отношение к таутомерному катализу в ферментах. Так, Уонг [55] высказал предположение, что перенос протона вдоль жестко фиксированной цепочки водородных связей в комплексе ES является неотъемлемой частью ферментативного катализа. Легко представить, что аналогичный синхронный сдвиг протонов может иметь место в связанных друг с другом карбоновой кислоте, имидазольной и фосфатной группах:
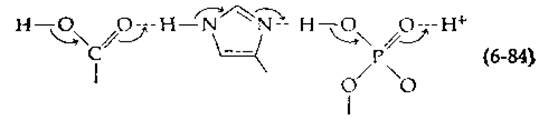
1) Динамике переноса протона в растворе с обсуждением биохимических модельных систем посвящен содержательный обзор Schuster Р., Wolschann Р, Tortschanoff К. в сборнике Molecular Biology, Biochemistry and Biophysics, v. 24, Chemical relaxation in molecular biology, Pecht I. and Rigler R. (Eds), Berlin, Springer-Verlag, 1977, pp. 107—109. — Прим. пepeв.
2) Частота колебаний ОН-связи равна частоте инфракрасного света, возбуждающего эти колебания. Частота v равна произведению волнового числа (для —ОН-связи оно составляет 3710 см-1) на скорость света с (3∙1010см∙с-1). Следовательно, частота колебаний —ОН-связи в молекуле воды v = 3∙1010∙3710 = 1,14∙1014 с-1.
В результате происходит перенос протона от одного конца цепи к другому [как и на схеме (6-83)], что обеспечивается легким протеканием реакций таутомеризации. Аналогичный процесс может идти и при участии боковых групп белка, объединяя две группы активного центра и способствуя таким образом согласованному кислотно-основному катализу, подобно тому, как это происходит на схеме (6-79).
В белках возможны и другие процессы таутомеризации (при наличии менее стабильных «минорных таутомеров»). Так, например, может иметь место сдвиг следующего типа с участием пептидных связей в а-спирали или ß-структуре:
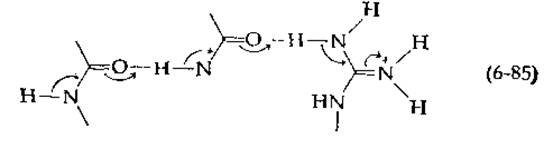
Как показано на схеме, электроны перемещаются к гуанидиновой группе боковой цепи аргинина. Можно рассмотреть и другие варианты (и таких вариантов множество, особенно если учесть, что в реакции могут принимать также участие коферменты или пуриновые и пиримидиновые основания). Все подобные процессы протекают исключительно быстро и с трудом регистрируются.
з. Влияние pH на активность ферментов
Изложенные выше данные об участии кислотных и основных групп: в ферментативном катализе получены в основном в результате исследований неферментативных модельных реакций. Имеются ли какие-либо указания на то, что ферменты действительно содержат подобные группы? Да, имеются, и наиболее четкое из них — зависимость ферментативной активности от pH. Довольно часто кривая зависимости Vmах от pH имеет колоколообразную форму (рис. 6-13). Оптимальное значение Vmах наблюдается при значении pH, часто лежащем в интервале от 6 до 9. Кривые колоколообразной формы проще всего объяснить, допустив, что в активном центре фермента имеются две ионогенные группы — а и b. В этом случае фермент может находиться в трех формах, различающихся по степени протонирования, — Е, ЕН, ЕН2:
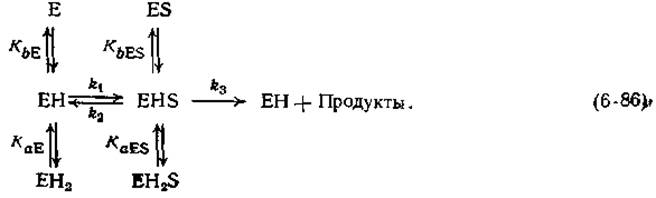
Обозначим константы диссоциации ДЛЯ двух групп в ферменте через КаЕ и КbE, а соответствующие значения для комплекса ES — через KaESи KbES. Константы скорости k1, k2и k3характеризуют стадии образования и распада комплекса ES.
Если допустить, что единственной реакционноспособной формой фермента, распадающейся с образованием продуктов, является форма EHS, то зависимости максимальной скорости от pH будут иметь колоколообразную форму (рис. 6-13). Подобные кривые часто встречаются в ферментативной кинетике, что подтверждает адекватность схемы (6-86). Кроме того, если для протекания реакции группа а фермента должна быть диссоциирована до сопряженного основания, а группа b должна находиться в протонированном состоянии, то естественно допустить, что эти две группы участвуют в кислотно-основном катализе.

РИС. 6-13 Теоретические зависимости Vmах от pH, рассчитанные исходя из уравнения (6-87) при k3[E]t = 1, pKaES = 6 и следующих значениях pKbES: I — 7, II — 8 и III —10 [58] Построенные при помощи ЭВМ графики любезно предоставлены К. Харрисом (С. Harris).
Для простого случая, описываемого схемой (6-86), рН-зависимости максимальной скорости и константы Михаэлиса описываются соответственно следующими выражениями:
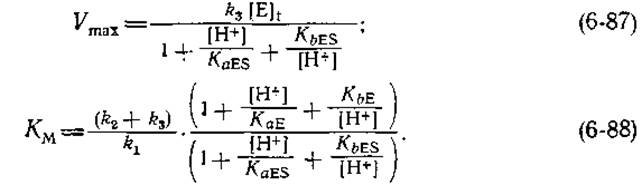
Когда фермент полностью насыщен субстратом, присутствуют только формы EH2S, EHS и ES. Из определения KaESи КbES следует, что при этом выполняется соотношение
![]()
В скобках стоит та же величина, что и в знаменателе выражений (6-87) и (6-88); иногда ее называют рН-функцией Михаэлиса [56]. Аналогичная по виду pH-функция для свободного фермента стоит в числителе выражения (6-88). pH-зависимость ферментативной активности часто имеет более сложный вид, чем зависимости, представленные на рис. 6-13 и описываемые приведенными выше уравнениям. Однако не составляет труда записать pH-функции Михаэлиса, а также соответствующие уравнения, подобные уравнениям (6-86) - (6-89), и для фермента с любым числом ионогенных групп в формах Е и ES. Следу иметь в виду, что в том случае, когда свободный субстрат содержит группы, диссоциирующие в том интервале pH, в котором изучается ферментативная активность, в числителе выражения (6-88) появится и рH-функция для свободного субстрата. Если в изменение активности фермента при варьировании pH вносит вклад изменение конформации белковой молекулы, то возможно кооперативное присоединение или отдача более чем одного протона, что должно отразиться на рН-функции Михаэлиса. Иногда это может привести к появлению дополнительного члена, аналогичного (4-32). Характер pH-зависимости ферментативной активности был детально проанализирован Диксоном [56], который предложил строить графики зависимости lg Vmах (или логарифма удельной активности) и — lg КM от pH.
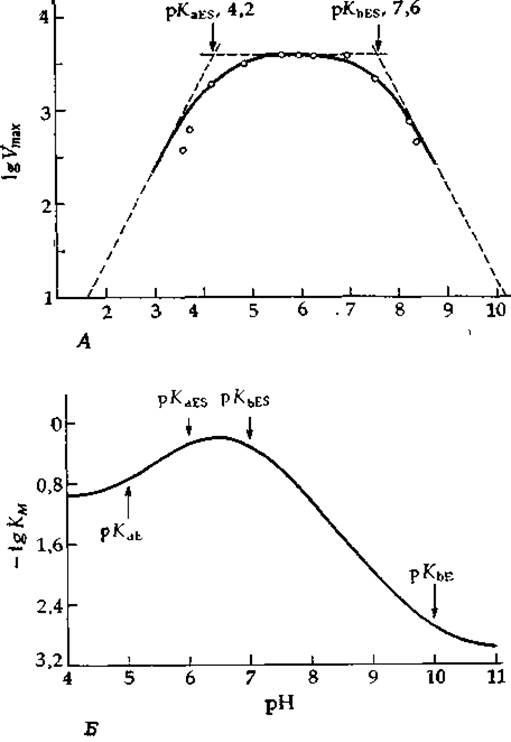
РИС. 6-14. А Зависимость Vmах от pH для кристаллической бактериальной а-амилазы [56а]. Б. Теоретическая зависимость lg КM от pH, рассчитанная исходя из уравнения (6-88) при рKаЕ = 5, рKbЕ = 10, pKaES = 6 и pKbES = 7. Построенный при помощи ЭВМ график любезно предоставлен К. Харрисом.
Типичные кривые такого рода представлены на рис. 6-14. В области значений рН = рKа± ~ 1,5 график зависимости lg Vmах от pH имеет криволинейную форму, однако за пределами этого интервала ветви кривой асимптотически приближаются к прямым, наклон которых равен единице, если в процессе диссоциации участвует один протон, или больше единицы, если имеет место кооперативная диссоциация нескольких протонов. Точки пересечения асимптот с отрезком прямой, параллельной оси абсцисс (ом. рис. 6-14), соответствуют значениям рKа. (Следует, однако, отметить, что к более надежным результатам приводит описание всех экспериментальных точек единой теоретической кривой.) Криволинейный участок проходит всегда ниже (или выше) точек пересечения на величину, равную lg 2 = 0,30 (или меньшую, если диссоциация протонов происходит кооперативно).
Вогнутые участки кривых зависимости lg KM от pH дают значения рKа свободного фермента или свободного субстрата, а выпуклые — значения рKа комплекса ES. Подобный подход к анализу рН-зависимостей параметров ферментативной реакции широко применяется энзимологами, однако он часто приводит к некорректным результатам. Например, многие кривые такого рода имеют очень резкий перегиб, криволинейный участок у них занимает интервал рН<3; при этом расстояние по ординате между кривой и точкой пересечения асимптоты с горизонтальным участком гораздо меньше 0,301). Это означает, что связывание протонов происходит кооперативно и кажущийся рKа сходен в таком случае с константой K в уравнении (4-33). Читателю, желающему глубже разобраться в обсуждаемых вопросах, можно рекомендовать теоретическую работу [57], где рассматриваются значения рKа переходного состояния. Интересный анализ кривых с необычно острым максимумом проведен в работах [57а,b].
Примером фермента, детально изученного с точки зрения влияния pH на кинетические параметры, может служить фумараза — фермент, катализирующий обратимую гидратацию фумаровой кислоты до яблочной [схема (6-64)]. В своей ранней очень интересной работе Алберти и др. [58] показали, что колоколообразная pH-зависимость имеет место как для прямой, так и для обратной реакции. Используя уравнения (6-88) и (6-89), эти исследователи рассчитали кажущиеся значения рKа для групп а и b фермента в буферных растворах с ионной силой 0,01 (табл. 6-1). Важно отдавать себе отчет в том, что кинетика этой обратимой реакции описывается более сложными уравнениями, чем уравнения (6-87)—(6-89), и поэтому кажущиеся значения рKа могут не совпадать с истинными. Однако очень заманчиво было бы допустить, что два значения рKа для свободного фермента, равные 6,2 и 6,8, соответствуют идентичным группам, по-видимому, имидазольным, со значениями микроскопических рKа, составляющими — 6,5. Свойства фумаразы будут обсуждаться далее в гл. 7, разд. 3,6.
Мутаротация глюкозы у Е. coli катализируется специфической мутаротазой [59], имеющей число оборотов 104 с-1. Форма графика зависимости — lg KM от pH указывает на наличие в свободном ферменте двух ионогенных групп со значениями рКa 5,5 и 7,6, а из характера зависимости lg Vmах от pH следует, что в комплексе ES присутствует одна группа с рКа = 4,75 [59]. Последняя может представлять собой каталитическую группу [группу В' в схеме (6-79)], возможно имидазол в форме сопряженного основания. Почему группа, имеющая в свободном ферменте рKа = 7,6, никак не проявляется в комплексе ES? Либо эта группа не участвует в катализе, либо величина рКа для нее так сильно сдвигается при связывании субстрата, что группу не удается выявить с помощью кривой зависимости lg Vmax от pH. Таким образом, экспериментальные данные не позволяют окончательно решить вопрос о том, участвует ли в работе мутаротазы ионогенная группа, соответствующая на схеме (6-79) группе —ВН+.
1) Удивительно, что для большей части кривых, приводимых Диксоном и Уэббом [56] для иллюстрации pH-профилей ферментативной активности, наблюдается такое же отклонение от теоретической зависимости Многие из полученных значений рKа могут быть некорректными из-за участия в процессе диссоциации нескольких протонов, что приводит к более резким pH-переходам, или в результате экспериментальных ошибок (например, ошибок, связанных с влиянием буфера).
Таблица 6-1 Кажущиеся значения рКa для фумаразы и ее комплексов с фумаратом и малатом [58]
|
Свободный фермент |
Kомплекс фермента с фумаратом |
Комплекс фермента с малатом (в случае обратной реакции) |
|
|
pК |
6,2 |
5,3 |
6,6 |
|
pKb |
6,8 |
7,3 |
8,5 |