Биохимия - Химические реакции в живой клетке Том 3 - Д. Мецлер 1980
Метаболизм азотсодержащих соединений
Серин и глицин
Порфобилиноген, порфирины и родственные соединения
В 1946 г. Шемин и Риттенберг [73а] описали один из первых примеров успешного использования радиоактивных меток для изучения метаболизма. В этой классической работе было продемонстрировано, что атомы порфиринового кольца в молекуле гема происходят из таких простых соединений, как ацетат и глицин. Как мы теперь знаем, ацетат в цикле трикарбоновых кислот превращается в сукцинил-СоА. В митохондриальном матриксе животных клеток сукцинил-СоА конденсируется с глицином, образуя б-амннолевулнновую кислоту [уравнение (8-20)] [73b], которая далее превращается в порфобилнноген (гл. 10, разд. Б, 1), непосредственный предшественник порфиринов. Путем деструкции 14С-порфиринов, образовавшихся из меченых молекул ацетата и глицина, Шемин и Риттенберг установили ту расстановку изотопных меток в пиррольном ядре, какая указана на рис. 14-13 для порфобилиногена. Черными кружками показаны те атомы, которые первоначально были Углеродными атомами метальной группы в молекуле ацетата (не следует забывать, что ацетильные группы ацетил-СоА проходят через цикл трикарбоновых кислот более одного раза и метка попадает из метальных групп ацетата как во 2-е, так и в 3-е положения сукцинил-СоА). Атомы, отмеченные белыми кружками, в основном происходят от мерильного углерода ацетата и в меньшей степени от карбоксильного углерода. Атомы, отмеченные звездочками, произошли от глицина, а непомеченные атомы углерода — от карбоксильного углерода ацетата.
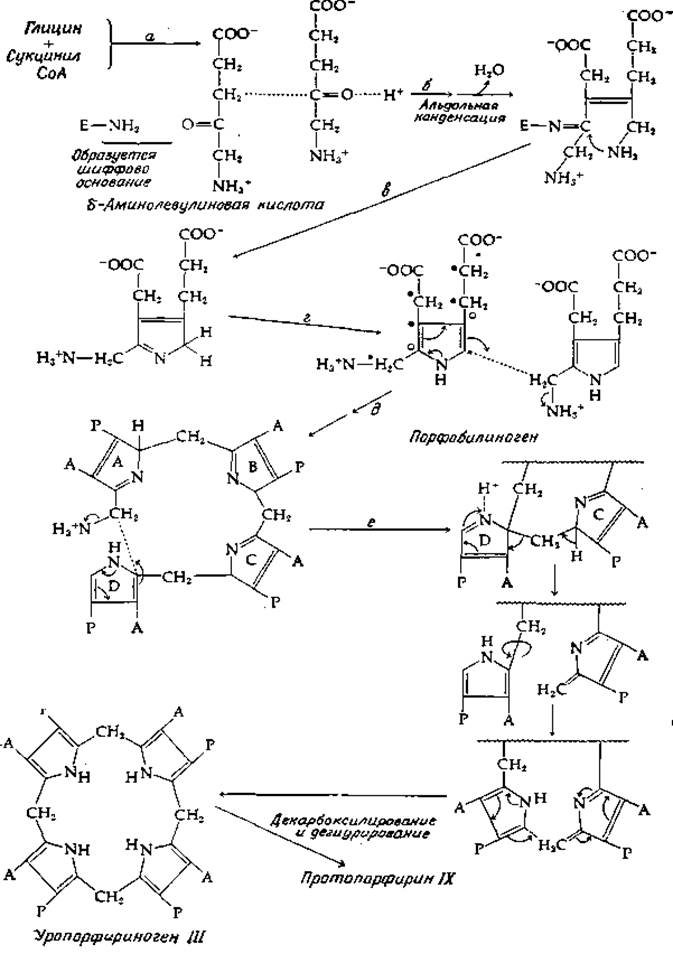
РИС. 14-13. Биосинтез порфиринов из глицина и сукцинил-СоА.
В растениях используется иной путь. В δ-аминолевулинат входит целиком весь 5-углеродный скелет а-кетоглутарата. Возможно, процесс начинается с внутримолекулярной окислительно-восстановительной реакции (возможно, после предварительного превращения а-кетоглутарата в тиоэфир), похожей на идущую в обратном направлении реакцию, катализируемую глиоксилазой I (гл. 7, разд. Л). Продуктом этой реакции должен быть у,δ-диоксовалерат который подвергается восстановительному аминированию с образованием 6-аминолевулината [74].
![]()
Как показано на рис. 14-13, превращение двух молекул δ-аминолевулиновой кислоты в порфобилиноген является многостадийной реакцией, начинающейся с альдольной конденсации (стадия б). Полагают, что фермент, катализирующий эту реакцию, образует шиффово основание с карбонильной группой одной из молекул субстрата, как указано на рисунке [75, 76]. За альдольной конденсацией следует дегидратация с образованием двойной связи между двумя атомами углерода, а замыкание кольца (стадия в) происходит путем последовательных перемещений иминной связи, подобных тем, которые были описаны ранее (т. II, стр. 228). Наконец, для образования порфобилиногена еще требуется таутомеризация (стадия г). Конденсация молекул порфобилиногена, приводящая к образованию порфиринов, протекает с участием двух ферментов, порфобилиногендезаминазы и уропорфириноген ІІІ-косинтетазы. Дезаминаза катализирует стадию д (рис. 14-13). Аммиак в результате элиминируется, но это необязательно происходит путем показанной на рисунке реакции прямого замещения. Может наблюдаться и перемещение электрона из соседнего азота в том же кольце. Нетрудно представить себе четырехкратное повторение такой конденсации с образованием симметричного предшественника молекулы уропорфирина I (рис. 10-1). В присутствии косинтетазы протекает реакция иного рода. Обратите внимание, что пятичленное кольцо в порфобилиногене характеризуется симметричным расположением двойных связей. Таким образом, реакция конденсации может затрагивать любой из атомов, находящийся в a-положении относительно кольцевого азота. За серией реакций конденсации (рис. 14-13, стадия е) следуют таутомеризация, разрыв и воссоединение цикла таким образом, чтобы порядок расположения карбоксиметильных и карбоксиэтильных боковых цепей отвечал уропорфириногену III. Последующий ряд реакций декарбоксилирования и окисления [77] ведет прямо к образованию протопорфирина IX.
Включение в протопорфирин ферро-иона требует участия специального фермента, протогем — ферролиазы (феррохелатазы) [78, 79]. Установлено, что этот фермент прочно связан с внутренней мембраной митохондрий животных клеток, хлоропластов растений и хроматофоров бактерий. Хотя обычно Fe2+, по-видимому, является единственным ионом металла, включающимся в порфирин, в дрожжах в заметных количествах накапливается [Zn2+]-протопорфириновый хелат; известен также комплекс с Сu2+ (гл. 10, разд. Б, 1).
а. Хлорофилл [80, 81]
Первой стадией превращения протопорфирина IX в хлорофилл может быть включение Mg2+. Эта реакция самопроизвольно почти не идет и требует участия катализаторов. Далее происходит метилирование карбоксиэтильной боковой цепи кольца III [уравнение (14-35)].

Остальные стадии образования хлорофилла представляют собой насыщение винильной группы при кольце IV, замыкание кольца V и присоединение остатка фитила (см. рис. 13-19, на котором представлены структуры хлорофиллов). Замыкание кольца V происходит вслед за ß-окислением трехуглеродной боковой цепи, как показано в уравнении (14-36). За этим следует окислительное замыкание кольца в протохлорофиллид а.
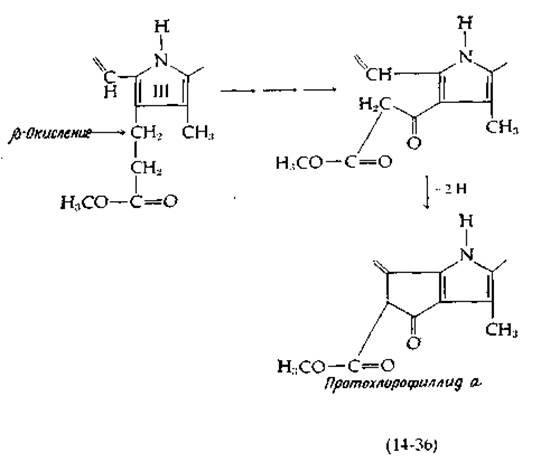
Последний соединяется с фитолом (вероятно, с промежуточным образованием фитилпирофосфата), в результате чего образуется хлорофилл а. Хлорофилл b, по всей вероятности, получается из хлорофилла а, а бактериохлорофиллы синтезируются из хлорофиллида а, причем фитильные группы присоединяются уже после восстановления кольца IV.
б. Коррины
Образование витамина В12 и других корринов требует сжатия цикла с элиминированием метенового мостика между кольцами А и D порфиринов (дополнение 8-Л). Естественно предположить, что метильная группа при атоме С-1 корринового кольца образуется из того же атома углерода, что и метеновый мостик в порфиринах (нетрудно представить себе такую модифицированную реакцию конденсации, при которой замыкание кольца на стадии е (рис. 14-13) происходило бы путем нуклеофильного присоединения к связи C = N кольца А). Однако данные 13С-ЯМР исключают такую возможность. Когда синтез витамина B12 проводили в присутствии 13С-метилсодержащего метионина, исследование продукта показало, что 13С попал в семь метильных групп. Мечеными оказались все «лишние» метильные группы, размещающиеся вдоль периферии молекулы, а также при атоме С-1 [82]. С помощью других экспериментов было установлено, что уропорфириноген III является предшественником витамина В12. Из этого следует, что цикл должен сначала замкнуться обычным способом, а затем снова раскрыться между кольцами А и D — с удалением углерода, образующего метиленовый мостик [83]. Были предложены и альтернативные механизмы [83а].
Дополнение 14-В
Метаболизм железа
Железо является одним из элементов, наиболее распространенных в земной коре; в обычных почвах его содержание достигает ~4%. Функции железа в живых клетках многочисленны и разнообразныа-в. Общее содержание железа в бактериях и грибах составляет в среднем ~1 ммоль/кг, но в тканях животных его, как правило, меньше. 70% из 3—5 г железа, содержащихся в организме человека, сосредоточено в эритроцитах, где общее содержание железа составляет ~20 мМ. В остальных тканях общее содержание железа составляет лишь ~0,3 мМ; в основном оно приходится на разного рода резервные формы. Суммарное содержание всех железосодержащих ферментов составляет — 0,01 мМ. Хотя средние концентрации получаются низкими, железо сконцентрировано в окислительных ферментах в мембранах, и, следовательно, локальные его концентрации могут быть значительно выше. Удивительно, что одна из групп анаэробных бактерий, а именно молочнокислые бактерии, которые вообще не содержит ферментов, реагирующих с кислородом, по всей видимости, полностью лишена и железа, и меди. Во всех других организмах железо обязательно должно присутствовать.
В тканях человека и других животных, а также в зеленых растениях и грибах значительная часть железа находится в форме ферритина, красновато-коричневого водорастворимого белкаг-е. Ферритин представляет собой резервную форму Fe (ІІІ) в растворимом и нетоксичном состоянии, легко пригодном для использования. Ферритин — несколько необычный белок. Содержание железа в ферритине составляет 17—23%, причем оно находится в виде расположенной в центре плотной массы гидратированной гидроокиси железа (III), заполняющей пространство диаметром 7 нм. Эта масса окружена белковой оболочкой из 24 субъединиц, расположенных в соответствии с кубической симметрией, во многом аналогично тому, как это показано на рис. 8-17. Внешний диаметр частицы составляет ~12 нм. Молекулярный вес апоферритина равен 445 000, а каждая субъединица имеет молекулярный вес 18 500. Полностью заполненная (до 23% Fe) молекула ферритина содержит свыше 2000 атомов железа, упакованных почти как в кристаллической решетке. Сердцевина молекулы легко различима в электронном микроскопе, и в микроскопии ферритин часто используют как своеобразный маркер. Другая резервная форма железа, гемосидерин, по-видимому, состоит из молекул ферритина вместе с добавочным количеством железа. При введении в организм избыточного количества железа отложение гомосидерина, в печени может дойти до токсического уровня.
Трудная проблема возникает у всех организмов в связи с относительной нерастворимостью гидроокиси железа (III) и других соединений, играющих роль резерва железа. В связи с этим железо часто захватывается в хелатной форме и переносится от одного органического лиганда (чаще всего белка) к другому, почти не проходя через состояние свободного Fe3+. Типичные константы образования хелатов Fe2+ лежат между значениями, свойственными комплексам Мn2+ и Со2+ (гл. 4, разд. В, 8, б; табл. 4-2). Например, для Fе2+-хелата ЭДТА logK1 = 14,3. Как и следовало ожидать, железо в форме Fe3+, имеющее меньшие размеры и более высокий заряд, связывается более прочно (log К1 = 25,0). Важное биохимическое значение имеет тот факт, что Fe3+ оказывает предпочтение кислородсодержащим лигандам.
С другой стороны, Fe2+ предпочтительнее связывается с азотом.
Важно также и то, что Fe3+, связанный с кислородными лигандами, легко обменивается на другие присутствующие в cреде ферри-ионы, тогда как связанный с азотсодержащими лигандами, в частности с гемом, Fe3+ обменивается очень медленно. Это свойство может иметь большое значение для соединений, транспортирующих железо, и для железосодержащих ферментов.
Если во внешней среде поддерживается достаточно высокая концентрация железа (например, для Е. coli 50 мкМ и более), то бактерии и другие микроорганизмы поглощают это железо без затруднений. Однако при низких концентрациях железа во внешней среде для повышения растворимости железа и переноса его внутрь клетки используются специальные соединения (сидерохромы)ж. Так, при концентрациях железа 2мкМ или ниже Е. coli и родственные кишечные бактерии выделяют в среду большие количества специфического хелатирующего агента энтеробактина (рис. 2-39). Очень устойчивый комплекс этого соединения с Fe3+ переносится внутрь бактериальной клетки специальной транспортирующей системой. Внутри клетки энтеробактин разрушается эстеразой, и диссоциация железа облегчается его восстановлением в Fe2+. Ряд сидерохромов содержит в центрах связывания железа гидроксаматные группы
![]()
К ним, в частности, относится пептид феррохром (вырабатываемый некоторыми бациллами). Отметим, что и в этих соединениях связи с железом образует кислород. В феррихроме Fe3+ связан очень прочноз; log К для образования комплекса из Fe3+ и свободного тригидроксаматного лиганда равен 29.
В среднем суточный рацион человека содержит ~15 мг железа; из них в организме всасывается около 1 мг. Обычно этого достаточно, чтобы компенсировать небольшие потери железа, в основном с желчью. В организме человека нет, по-видимому, механизма, обеспечивающего выведение из организма избыточных количеств железа; содержание железа регулируется только уровнем его поступления в организм. Этот уровень повышается у женщин во время беременности и у молодых женщин при менструальном кровотечении (для возмещения потери железа с кровью). Избыточные количества железа могут быть сильно токсичны. Механизм регуляции всасывания железа пока неясен, но установлено, что, оказавшись в организме, Fe3+ связывается трансферрином — белком с мол. весом 80 000, содержащим два центра связывания железаи. Вместе с каждым ионом Fe3+ связывается сопутствующий анион. Трансферрин цыплят, по-видимому, идентичен железосвязывающему белку кональбумину, содержащемуся в белке куриных яиц. С другой стороны, лактоферрин, красный железосвязывающий белок, присутствующий в молоке, отличается от трансферрина крови своей аминокислотной последовательностью. Железосвязывающие белки, находящиеся в жидкостях тела, иногда объединяют в общую группу с общим названием сидерофилины.
Главной функцией трансферрина является транспорт железа в организме, но он может служить и буфером, регулирующим поступление железа; возможно, всасывание железа через слизистую кишечника регулируется степенью насыщения железом трансферрина в крови. Для переноса железа из трансферрина в гем, происходящего в юных клетках крови, образующихся в костном мозге, Fe3+ должен восстановиться в Fe2+. Восстановление ферри-иона, вероятно, необходимо и для освобождения его из ферритинак. Механизмы этих процессов не установлены, но известно, что в роли восстановителей могут выступать аскорбиновая кислота или глутатион. Ферри-ион в гемоглобине (метгемоглобине) восстанавливается NADH-зависимым ферментом (дополнение 10-А). Наряду; с этим Fe2+, видимо, должен иногда подвергаться окислению в Fe3+ под действием медьсодержащей ферроксидазы (церулоплазмина; дополнение 10-3). Попав в организм, железо тщательно в нем удерживается. Так, в результате ежесуточного разрушения 9 млрд. эритроцитов освобождаются 20— 25 мг железа, которое почти все снова используется или резервируется в организме.
а Neilands J. В., ed., Microbial Iron Metabolism, Academoc Press, New York, 1974.
б Jacobs A., Worwood M., eds., Iron in Biochemistry and Medicine, Academic Press, New York, 1974.
в O'Dell B. L, Campbell B. J., Compr. Biochem., 21, 179—265 (1970).
г Harrison P. M., Hoy T. G., in: Inorganic Biochemistry (G. L. Eichhorn, ed.), Vol. 1, pp. 253—279, Elsevier, Amsterdam, 1973.
д Hoare R. J., Harrison P. M., Hoy T. G., Nature (London), 255, 653—654 (1975).
e Massover W. H., Cowley J. M., PNAS, 70, 3847—3851 (1973).
ж Neilands J. B., in: Inorganic Biochemistry (G. L. Eichhorn, ed.), Vol. 1, pp. 167—2020, Elsevier, Amsterdam, 1973.
з Rosenberg H., Young I. G., in: Microbiol Iron Metabolism (J. B. Neilands, ed.), pp. 67—82, Academic Press, New York, 1974.
и Aisen P., in: Inorganic Biochemistry (G. L. Eichhorn, ed.), Vol. 1, pp. 280— 305, Elsevier, Amsterdam, 1973.
к Cavill I., Worwood M., Jacobs A., Nature (London), 256, 328—329 (1975).
в. Порфирия [84—86]
Организм человека использует не весь образующийся порфобилиноген; в норме небольшие его количества обычно выводятся с мочой, главным образом в виде копропорфиринов (гл. 10, разд. Б, 1). Существуют наследственные и приобретенные нарушения, при которых содержание порфиринов в крови повышено и с мочой выделяются значительно большие их количества (порфирия). Бывают случаи, когда порфирия протекает в легкой форме и почти не сопровождается какими-либо симптомами, но в других случаях в коже под роговым слоем откладываются интенсивно флуоресцирующие свободные порфирины, что сопровождается фотосенсибилизацией и приводит к изъязвлению кожи. В наиболее тяжелых случаях экскретируемые порфирины придают моче винно-красный цвет. У больных развиваются тяжелые неврологические поражения. Наблюдается и целый ряд других симптомов1). При одной форме врожденной порфирии с мочой выделяются большие количества уропорфирина I. Биохимический дефект в этом случае, по-видимому, сводится к недостаточному синтезу косинтетазы, необходимой для образования протопорфирина IX. Другая форма порфирии обусловлена образованием в печени избыточных количеств δ-аминолевулиновой кислоты. Существует предположение, что лечить таких больных, возможно, следует введением бензоата или n-аминобензоата [87]. Смысл такого воздействия состоит в том, чтобы переключить обмен глицина на синтез гиппуровой кислоты (дополнение 9-А) или ее n-аминопроизводного, снижая тем самым скорость синтеза порфиринов.
При некоторых легких формах порфирин прием определенных лекарственных препаратов может вызвать острый приступ болезни. Медикаменты и другие химикалии иногда вызывают порфирию в результате индукции чрезмерного синтеза δ-аминолевулииат — синтетазы. Среди соединений, дающих такой эффект, можно назвать гексахлорбензол и тетрахлордибензодиоксин. Последний является одним из самых сильных известных агентов, индуцирующих образование синтетазы [88].
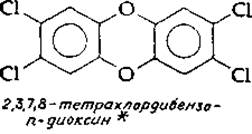
1) Яркое описание проявлений этой болезни, которые, как полагают, наблюдались у английского короля Георга III, дают Макэлпайн и Хантер [84].
* Нередкое присутствие этого диоксина в виде примеси в гербициде 2,4,5-трихлорофепоксиуксусной кислоте (2,4,5-Т) вызвало большую озабоченность. Вполне возможно, что рассматриваемый диоксин представляет собой наиболее токсичную среди всех известных малых молекул: при поступлении в организм с пищей LD50 этого соединения для морских свинок составляет всего 1 мкг на 1 кг веса [88]. К тому же он является сильным тератогенным агентом (вызывающим аномалии развития плода).
а. Желчные пигменты
Ферментативное разрушение гема представляет собой важный метаболический процесс уже хотя бы потому, что при этом освобождается железо, вновь используемое организмом. Некоторые из путей катаболизма гема показаны на рис. 14-14. Считается, что оксигенация (гидроксилирование) затрагивает сперва а-метеновый углерод (между кольцами А и В). Гидроксилированный продукт расщепляется с освобождением окиси углерода. Реакции катализируются микросомными гидроксилазами [89—90а]. В опытах с использованием 18О2 было показано, что образующийся дециклизованный тетрапиррол биливердин содержит два атома 18О, а СО2 — один атом І8О. Реакции восстановления и окисления биливердина приводят к образованию большого числа различных нециклизованных тетрапирролов.
Биливердин, первый продукт размыкания цикла, восстанавливается в билирубин, который транспортируется в печень в виде комплекса с сывороточным альбумином. В печени билирубин превращается в глюкурониды [уравнение (12-12)], образующиеся за счет гликозилирования боковых остатков пропионовой кислоты. Многие из конъюгатов билирубина поступают в желчь. В кишечнике они вновь гидролизуются до свободного билирубина, который восстанавливается кишечными бактериями в уробилиноген, стеркобилиноген и мезобилирубиноген. Эти соединения бесцветны, но легко окисляются кислородом в уробилин и стеркобилин. Некоторая часть уробилина и других желчных пигментов вновь поступает в кровь и выделяется с мочой, придавая ей всем известный, характерный желтый оттенок.
Наблюдаемый при желтухе желтый цвет кожи может быть обусловлен чрезмерным разрушением гема (например, в результате избыточного гемолиза), неспособностью печени к образованию конъюгатов билирубина или какими-либо факторами, препятствующими поступлению продуктов распада гема в кишечный тракт.
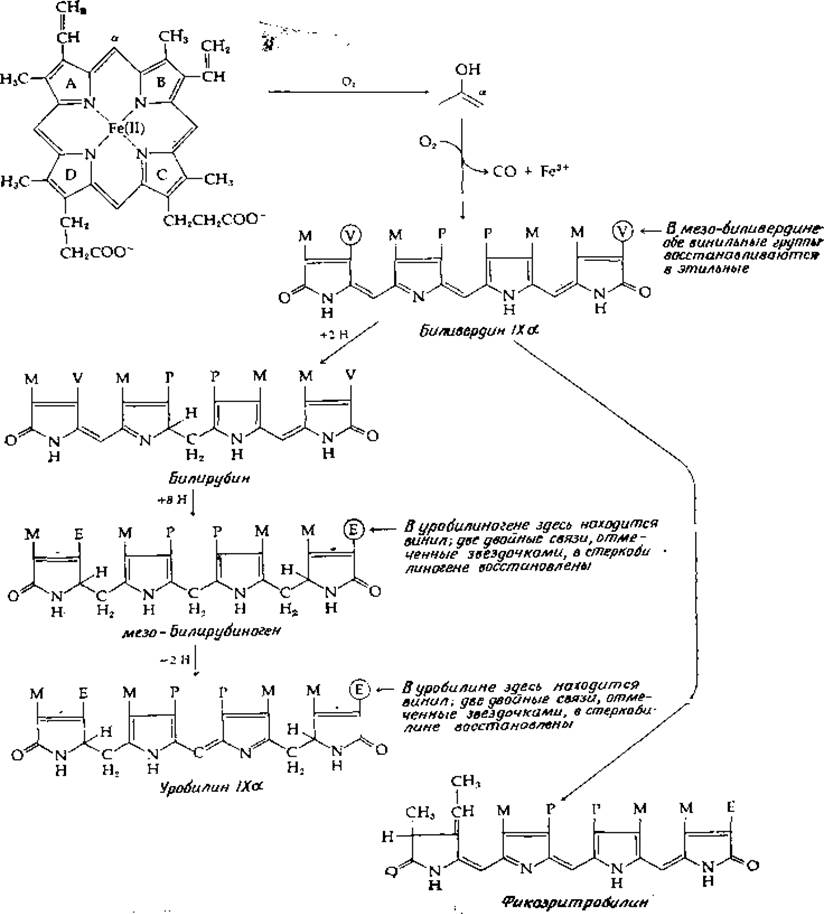
Об ациклических тетрапирролах водорослей и о хромофоре фитохрома мы уже упоминали (рис. 13-22). Все они происходят от фикоэритробилина, родственного биливердину, как это показано на рис. 14-14.