БИОХИМИЯ - Л. Страйер - 1984
ТОМ 1
ЧАСТЬ I. КОНФОРМАЦИЯ И ДИНАМИКА
ГЛАВА 7. МЕХАНИЗМ ДЕЙСТВИЯ ФЕРМЕНТОВ: ЛИЗОЦИМ И КАРБОКСИПЕПТИДАЗА
7.6. Промежуточное образование иона карбония - критический этап катализа
На основе изложенных структурных данных Филлипс (Phillips) и сотрудники детально разработали вероятный механизм каталитического действия лизоцима. В каталитическом цикле они выделили следующие важные этапы.
1. —СООН-группа остатка 35 передает Н+ на связь между С-1 кольца D и гликозидным атомом кислорода; в результате данная связь расщепляется (рис. 7.14).
Рис. 7.14. Первый этап постулированного механизма действия лизоцима -перенос Н + от Glu 35 на атом кислорода гликозидной связи. При этом происходит расщепление гликозидной связи и образование иона карбония
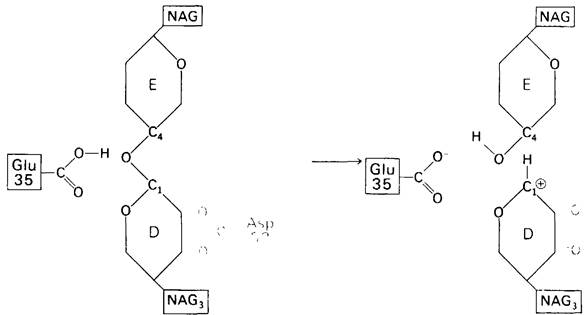
2. Это создает положительный заряд на С-1 кольца D. Образовавшийся короткоживущий продукт называется ионом карбонил, поскольку он содержит положительно заряженный атом углерода.
3. Димер NAG, состоящий из остатков Е и F, отдаляется от фермента в результате диффузии.
4. Образовавшийся в качестве промежуточного продукта ион карбония реагирует с ОН-- группой растворителя (рис. 7.1 5). Тетра-NAG, состоящий из остатков А, В, С и D, отдаляется от фермента путем диффузии.
Рис. 7.15. Реакция гидролиза завершается присоединением ОН к промежуточно образовавшемуся иону карбония и Н+ к боковой цепи Glu 35
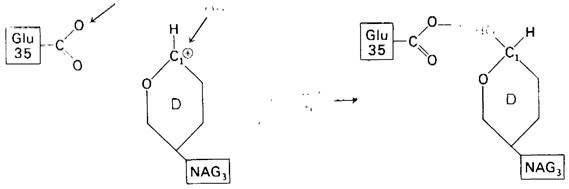
5. Глутамат-35 вновь протонируется, и фермент может вступать в новый каталитический цикл.
Основные элементы этой схемы катализа следующие.
1. Общий кислотный катализ. Источник протона-глутамат-35, который находится в деионизированной форме и отстоит от гликозидного атома кислорода на оптимальное расстояние-3 А.
2. Образование в качестве промежуточного продукта иона карбония. Два разных по своей природе фактора, оказывающих стабилизирующий эффект на ион карбония, значительно облегчают осуществление ферментативной реакции. К ним относятся:
а. Электростатический фактор, т.е. присутствие отрицательно заряженной группы поблизости (в 3 А) от образующегося в качестве промежуточного продукта иона карбония. Аспартат-52, находящийся в форме отрицательно заряженного карбоксилат-иона, электростатически взаимодействует с положительным зарядом на С-1 кольца D.
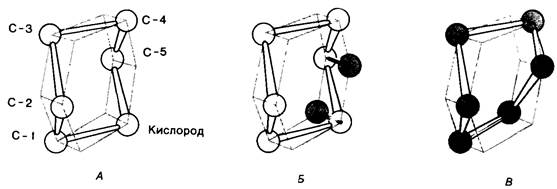
б. Геометрический фактор, а именно деформация кольца D (рис. 7.16). Гекса-NAG размещается наилучшим образом в щели активного центра фермента при условии, что геометрия кольца D меняется от конформации кресла к конформации полукресла. Такая деформация способствует ускорению катализа, так как при конформации полукрссла значительно облегчается образование иона карбонил. Расположение углеродных атомов 1,2 и 5 и кислородного атома кольца в одной и той же плоскости при конформации полукресла приводит к тому, что положительный заряд распределяется между С-1 и атомом кислорода кольца. Таким образом, связывая субстрат, фермент заставляет его принимать форму переходного состояния, а именно форму иона карбония.
Рис. 7.16. Изменение конформации кольца D субстрата лизоцима в конформацию полукресла. A-углеводный остаток в обычной конформации кресла; Б-при связывании с лизоцимом атом кислорода кольца и С-5 в углеводном остатке D перемещаются так, что С-1, С-2, С-5 и О оказываются в одной плоскости, как это показано на рис. В
7.7. Экспериментальное доказательство предложенного механизма ферментативного катализа
Основанная на рентгеноструктурных данных гипотеза о характере связывания субстрата и механизме катализа была проверена постановкой многочисленных химических экспериментов. Все полученные результаты подтверждают правильность выдвинутой гипотезы. Приведем ряд наиболее существенных экспериментальных доказательств.
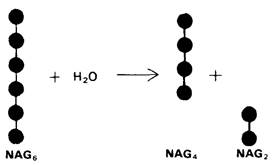
1. Способ расщепления субстрата. В соответствии с предположением о том, что расщепление гексамера происходит между 4-м и 5-м остатками, оказалось, что действительно гекса-NAG расщепляется на тетра-NAG и ди-NAG (рис. 7.17).
Рис. 7.17. Гекса-NAG гидролизуется с образованием тетра-NAG и ди-NAG
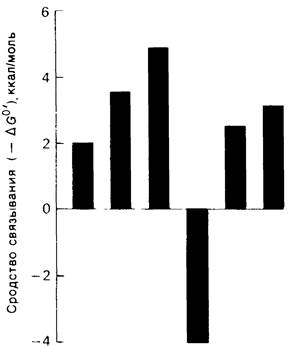
2. Сродство связывания. Путем измерения равновесия при реакции связывания с ферментом каждого из шести сахаров определяли их вклад в общую величину свободной энергии связей, представленных в гексамере (рис. 7.18). Обнаружилось поразительное явление: вклад углеводного остатка D был отрицательным. На связывание остатка D расходовалось около 4 ккал/моль. Этот результат подтверждает гипотезу о деформации остатка D при связывании с ферментом: изменение от конформации кресла к конформации полукресла требует энергии. Любопытно также, что остаток С вносит наибольший положительный вклад в сродство связывания. Действительно, рентгеноструктурные данные показывают, что остаток С образует большое число водородных связей и вандерваальсовых взаимодействий.
Рис. 7.18. Вклад каждого из шести углеводных остатков гекса-NАG в стандартную свободную энергию связывания этого субстрата. Связывание остатка и идет с потреблением энергии. Энергия расходуется на такое изменение формы остатка, чтобы он соответствовал активному центру фермента
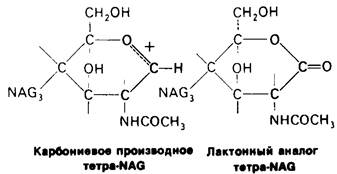
3. Аналоги переходного состояния. Представление о деформации углеводного остатка D в конформацию полукресла-важный аспект постулированного механизма катализа, поскольку конформация полукресла характерна для переходного состояния. Как уже отмечено выше, это представление подтверждается данными о затрате энергии на присоединение остатка D-энергии, расходуемой на деформацию. Другое подтверждение было получено при изучении аналога переходного состояния субстрата, т. е. соединения, имеющего как до, так и после связывания с ферментом такую же геометрию, как субстрат в переходном состоянии. Кольцо D лактонного аналога тетра-NAG (рис. 7.19) в кристалле тетрасахарида имеет конформацию полукресла. При связывании с лизоцимом атомы С-1, С-2, С-4, С-6 и кислородный атом кольца D этого аналога располагаются в одной плоскости. Такая конформация «софы» сходна с постулированной для переходного состояния конформацией полукресла; это означает, что лактонный аналог в отличие от тетра-NAG при связывании с лизоцимом деформируется очень мало. Оказалось, что лактонный аналог связывается с лизоцимом (в участках связывания от А до D) в 3600 раз сильнее, чем тетра-NAG. Отсюда вытекает, что при деформации кольца D нормального субстрата скорость расщепления может возрасти примерно в 3600 раз.
Рис. 7.19. Кольцо D лактонного аналога тетра-NAG имеет конформацию, похожую на полукресло, и в этом отношении сходно с промежуточным переходным состоянием в реакции, катализируемой лизоцимом
Роль этого фактора в катализе ясно предвидел Полинг (Pauling), о чем свидетельствует его лекция, читанная в 1948 г.:
«Я полагаю, что ферменты - это молекулы, которые по структуре комплементарны активированным комплексам тех реакций, которые они катализируют, т.е. молекулярной структуре, промежуточной между реагирующими веществами и продуктами реакции в данном каталитическом процессе. Сила притяжения молекулы фермента к активированному комплексу приводит к снижению энергии последнего, а, следовательно, к снижению энергии активации данной реакции и возрастанию скорости реакции».
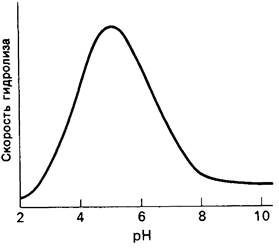
4. Зависимость скорости каталитической реакции от pH. Скорость гидролиза хитина достигает наиболее высоких значений при pH 5 (рис. 7.20). По обе стороны от этого оптимума ферментативная активность резко падает. Снижение активности при сдвиге pH в щелочную сторону обусловлено ионизацией глутамата-35, тогда как при сдвиге pH в кислую сторону - протонированием аспартата-52. Лизоцим проявляет ферментативную активность только при условии, что глутамат-35 находится в неионизированной форме, а аспартат-52 - в ионизированной.
Рис. 7.20. Скорость гидролиза хитина (роlу-NAG) лизоцимом как функция pH
5. Избирательная химическая модификация. Лизоцим сохраняет ферментативную активность, если все имеющиеся в нем карбоксильные группы, за исключением карбоксильных групп глутамата-35 и аспартата-52, подвергнуть этерификации. Остатки 35 и 52 остаются немодифицированными, если этерификацию проводят в присутствии субстрата. При удалении субстрата аспартат-52 этерифицируется (тогда как глутамат-35 остается неизменным). Модификация аспартата-52 приводит к полной инактивации фермента. Это подтверждает предположение о том, что, строгая ориентация карбоксилат-иона аспартата-52 необходима для стабилизации образующеюся в качестве промежуточного продукта иона карбония.
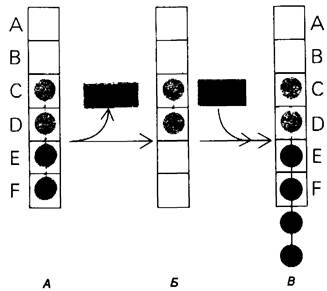
6. Трансгликозилирование. При добавлении к лизоциму тетра-NАG происходит медленное образование гекса-NАG и ди- NАG (рис. 7.21). Существование этой реакции, называемой трансгликозилированием, подтверждает наличие одного из важных элементов предложенного механизма ферментативной реакции, а именно образования в качестве промежуточного продукта комплекса гликозил — фермент.
Рис. 7.21. Существование промежуточного продукта гликозил- фермент подтверждается способностью лизоцима катализировать, хотя и медленно, реакцию трапа ликозилирования, NАG4 (показан красным) соединяется с промежуточным продуктом гликозил фермент (показан синим на рис. Б), и в результате образуется NAG6
При обычных гидролитических реакциях этот промежуточный продукт реагирует с ОН-. При трансгликозилировании в реакции используется вторая молекула углевода: ROH. Реакция трансгликозилирования специфична, так как акцептор связывается в участках Е и F в щели активного центра. Более того, образующаяся гликозидная связь имеет β-конфигурацию, как и в субстрате. Таким образом, эти данные подтверждают правильность предполагаемой формы промежуточного продукта катализа.