Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Динамика белков: от структуры к функционированию
Алгоритмы сэмплирования коллективных координат
TEE-REX
Улучшенные методы сэмплирования, такие как ОД (Amadei et al. 1996) достигают своей эффективности (Amadei et al. 1996; de Groot etal. 1996a, b) в основном благодаря тому, что в конфигурационной динамике белков преобладает лишь небольшое число внутренних коллективных степеней свободы. К тому же, системы, рассчитываемые с помощью таких методов, всегда находятся в неравновесном состоянии, что затрудняет расчет термодинамических, т.е. равновесных, свойств этих систем. С другой стороны, алгоритмы обобщенных ансамблей, такие как REX, не только улучшают сэмплирование, но и дают правильные статистические ансамбли, необходимые для расчета равновесных свойств, которые могут быть подвергнуты экспериментальной проверке. Однако REX быстро становится вычислительно невозможным для систем, имеющих больше, чем несколько тысяч частиц, что ограничивает его нынешнее применение расчетом небольших пептидов (Pitera and Swope 2003; Cecchini et al. 2004; Nguyen et al. 2005; Liu et al. 2005; Seibert et al. 2005). Новый алгоритм обмена репликами в коллективной динамике с улучшенной температурой TEE-REX (Temperature Enhanced Essential dynamics Replica Exchange) (Kubitzki and de Groot 2007) совмещает в себе привлекательные свойства REX со свойствами, возникающими из индивидуального возбуждения функционально значимых мод, не имея при этом недостатков обоих подходов.
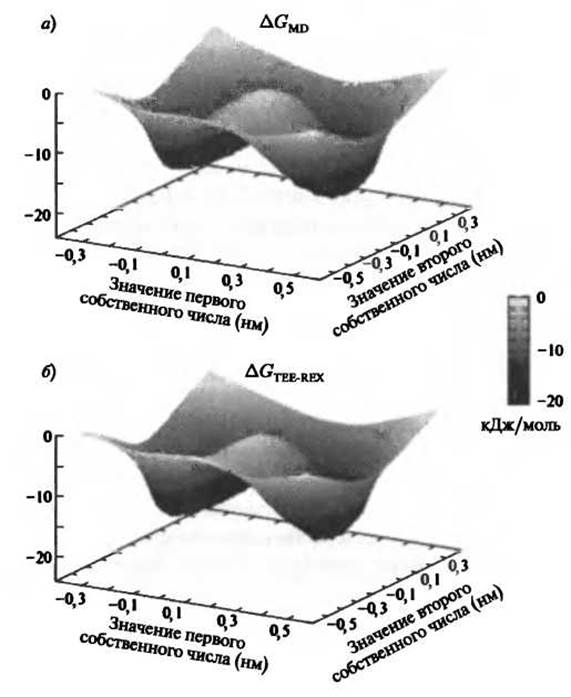
Рис. 9.9. Сравнение двумерных поверхностей свободной энергии (в кДж/моль) диаланина, вычисленная с помощью МД (панель а) и TEE-REX (панель б). Отклонение ∆GТЕЕ-REX - GMD соизмеримо со статистической ошибкой, составляющей -0.1 kβТ
На рисунке 9.6 показано схематическое сравнение алгоритма REX при стандартной температуре (слева) и алгоритма TEE-REX (справа). Алгоритм TEE-REX построен по той же схеме, что и REX: параллельно и независимо рассчитывается ряд реплик системы и периодически делаются попытки обмена между соседними репликами. Но в отличии от REX, во всех репликах, кроме референсной, термически возбуждаются только те степени свободы, которые дают значительный вклад в общие флуктуации системы (коллективное подпространство {es}). Это позволяет объединить несколько преимуществ при устранении недостатков. В отличие от стандартного алгоритма REX, индивидуальное возбуждение коллективных координат способствует сэмплированию по этим функционально важным модам движения, т.е. используется преимущество КД. Для компенсации недостатков, связанных с таким индивидуальным возбуждением, т.е. искажением ансамбля, схема встроена в протокол REX. В связи с этим получаемые ансамбли имеют приблизительно больцмановское распределение и используются улучшенные сэмплирующие свойства REX. Вероятность обмена (уравнение 9.1) между двумя репликами критически зависит от числа возбужденных степеней свободы системы. Поскольку такие степени свободы составляют лишь незначительную часть от общего числа степеней свободы системы, то обходится узкое место, которым являются низкие вероятности обмена в полноатомных расчетах по алгоритму REX. Таким образом, при заданных вероятностях обмена может быть использована настолько большая разница в температурах ∆Т, что потребуется лишь несколько реплик.
На рисунке 9.9 показана двумерная проекция ландшафта свободной энергии диаланина, вычисленная с помощью МД (панель А) и TEE-REX (панель В). Термодинамическое поведение системы можно считать полностью известным, если известен какой-нибудь термодинамический потенциал, такой как относительная свободная энергия Гиббса ∆G. Сравнение свободных энергий позволяет нам, таким образом, решить, до какой степени совпадают ансамбли, созданные разными вычислительными методами. При этом абсолютно необходимо, чтобы ансамбли имели идентичный состав. Для тестового случая диаланина это требование выполняется. Детальный анализ формы поверхностей свободной энергии, полученных из МД и TEE-REX, показывает, что максимальное абсолютное отклонение составляет 1.5 kJ/mol ≈ 0.6 квТ от идеального случая ∆GTEE-REX - GMD= 0, что соизмеримо с максимальной статистической ошибкой, составляющей 0.15 kвТ для каждого из методов. Небольшие отклонения, обнаруженные в ансамбле TEE-REX, вызваны, вероятно обменом неравновесных структур в референском ансамбле.
Для гуанилина, небольшого пептидного гормона, состоящего из 13 аминокислот, была оценена эффективность сэмплирования алгоритмом TEE-REX по сравнению с МД (Currie et al. 1992). Траектории, рассчитанные обоими методами при одинаковых затратах вычислительных ресурсов, были спроецированы на пространство (φ, ψ), а также на различные двумерные подпространства, которые задаются главными компонентами, вычисленными по МД ансамблю структур гуанилина. По этим проекциям было измерена временная эволюция сэмплированного объема конфигурационного пространства. В итоге оказалось, что производительность сэмплирования у МД довольно ограничена по сравнению с TEE-REX, который по этому показателю превосходит МД в среднем в 2,5 раза в зависимости от подпространства, использованного для проецирования.
9.3.2.1. Приложения: поиск переходного пути в аденилаткиназе
Понимание функциональных основ функционирования многих белков (Gerstein et al. 1994; Berg et al. 2002; Karplus and Gao 2004; Xu et al. 1997) требует подробного знания переходов между функционально значимыми конформациями. В последние годы рентгеновская кристаллография и спектроскопия ЯМР предоставляли в основном статические картины различных конформационных состояний белков, оставляя без ответа вопросы, касающиеся переходов между этими состояниями. Для расчетов МД атомарного разрешения прояснение путей и механизмов конформационной динамики белков представляет собой нетривиальную задачу из-за больших характерных времен. В этом отношении хорошим примером является аденилаткиназа из Е. coli. Это мономерный фермент, играющий ключевую роль в энергетическом балансе клетки, поскольку контролирует уровень АТФ в клетке, катализируя реакцию (Mg2+:ATФ) + АМФ ↔ (Mg2+:AДФ) + АДФ. Структура фермента состоит из трех доменов (Рис. 9.10): большого центрального домена “CORE” (показан светлосерым), АМФ-связывающего домена, называемого “AMPbd” (показан черным) и похожего на крышку АТФ-связывающего домена, называемого “LID” (показан темносерым), который закрывает фосфатные группы в активном центре (Müller et al. 1996). В отсутствии лиганда домены LID и AMPbd принимают открытую конформацию, если считать, что закрытая конформация наблюдается в структуре, закристаллизованной с ингибитором переходного состояния Ар5А (Müller and Schulz 1992). Здесь лиганды находятся в высокоспецифичном окружении, необходимом для катализа. Недавние ЯМР исследования по спиновой релаксации ядер 15N (Shapiro and Meirovitch 2006) показали существование движений каталитического домена в подвижных доменах AMPbd и LID в наносекундном диапазоне, в то время как релаксация домена CORE происходит в пикосекундном диапазоне (Tugarinov et al. 2002; Shapiro et al. 2002). Конформационная подвижность аденилаткиназы была рассмотрена в нескольких вычислительных работах (Temiz et al. 2004; Maragakis and Karplus 2005; Lou and Cukier 2006; Whitford et al. 2007; Snow et al. 2007) Однако из-за большой амплитуды движений и больших времен, на которых они происходят, в полноатомной МД до сих пор не удалось наблюдать самопроизвольных переходов между открытой и закрытой конформациями. Применение алгоритма TEE-REX облегчает наблюдение таких переходов и делает возможным получение полноатомного описания пути перехода и лежащих за ним механизмов (Kubitzki and de Groot 2008). Для осуществления такого описания из коротких МД расчетов произвольной конформации были сконструированы коллективные подпространства {es}, включившие в себя также структуры в открытой и закрытой конформации. В последнем случае моды {es}, включая разностную моду, связывающую открытые и закрытые экспериментальные структуры, были возбуждены.
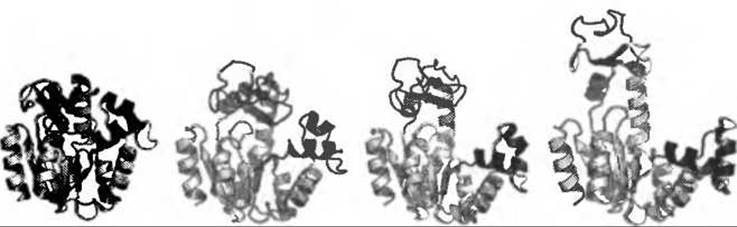
Рис. 9.10. Закрытая (слева) и открытая (справа) кристаллографические структуры аденилаткиназы из Е. coli вместе с промежуточными структурами, описывающими две фазы перехода “закрыто-открыто”. Аденилаткиназа состоит из домена CORE (показан светло-серым), домена AMPbd (показан черным) и домена LID (показан темно-серым). В закрытой структуре (слева) ингибитор переходного состояния Ар5А удален
Наблюдаемый путь перехода можно представить в виде двух фаз. Стартуя из закрытого состояния (Рис. 9.10 слева), домен LID остается закрытым, в то время как домен AMPbd, содержащий спирали а2 и а3, принимает полуоткрытую конформацию. В процессе этого спираль а2 изгибается к спирали а4 из домена CORE на 15 градусов по отношению к спирали а3. Такое открывание сайта связывания АМФ могло бы облегчить эффективное высвобождение образовавшегося продукта. Во время второй фазы наблюдается открывание домена L1D, частично скоррелированное с открыванием домена AMPbd. По сравнению с крупнозернистым подходом, полноатомный расчет по алгоритму TEE-REX делает возможным детальное рассмотрение взаимодействий между остатками. В случае аденилаткиназы это позволило выявить образование в первой фазе стабильного солевого мостика между остатками Asp118 и Lys136, связывающего домены LID и CORE. При оценке всех невалентных взаимодействий между этими доменами оказалось, что этот солевой мостик вносит значительный вклад во взаимодействие между доменами. Таким образом, разрыв мостика с помощью мутации, например, Asp118Ala должен уменьшить стабильность открытого состояния. При сравнении четырнадцати PDB-структур аденилаткиназы из дрожжей, кукурузы, человека и бактерий для одиннадцати из них оказался характерен такой солевой мостик на интерфейсе LID-CORE. Представляется возможным и альтернативный переходный путь, но анализ всех расчетов по алгоритму TEE-REX позволяет предположить наличие высокоэнергетического барьера, препятствующего полному открыванию домена AMPbd после того, как открывается домен LID. Наряду с наблюдаемыми значительными флуктуациями в элементах вторичной структуры, говорящими о высокой энергии внутренних деформаций, энтальпийные ограничения на этом пути, вероятно, делают его неподходящим на роль переходного пути в аденилаткиназе.