Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Динамика белков: от структуры к функционированию
Методы предсказания функциональных мод
CONCOORD
Программа CONCOORD (de Groot et al. 1997) использует геометрический подход к предсказанию подвижности белков. Пространственная структура белка определяется различными взаимодействиями, такими как ковалентные связи, водородные связи и неполярные взаимодействия. Большая часть этих взаимодействия остается неизменной во время функционально значимых конформационных перестроек. Это наблюдение лежит в основе метода CONCOORD: на основе данных об исходной структуре генерируются альтернативные структуры, в которых подавляющее большинство взаимодействий остается тем же, что и в исходной структуре. Для этого на первом шаге расчета методом CONCOORD анализируются взаимодействия в исходной структуре и на их основе создаются геометрические ограничения - в основном ограничения по расстоянию между атомами с верхней и нижней границами, а также ограничения по углам и информация о плоских и хиральных группах. Такое геометрическое описание структуры можно сравнить с планом сборки белка. На втором шаге, стартовав из случайного расположения атомов, структура итеративно перестраивается на основе ранее составленного плана - обычно это происходит несколько сотен раз. Поскольку каждый запуск стартует со случайного расположения атомов, то, в отличие от МД, этот метод не страдает от проблемы сэмплирования и приводит к получению ансамбля, охватывающего все конформационное пространство, которое доступно с учетом ранее полученных ограничений. Однако метод не дает информации ни о пути между двумя подсостояниями, ни о времени перехода, ни о его энергии (Рис. 9.12).
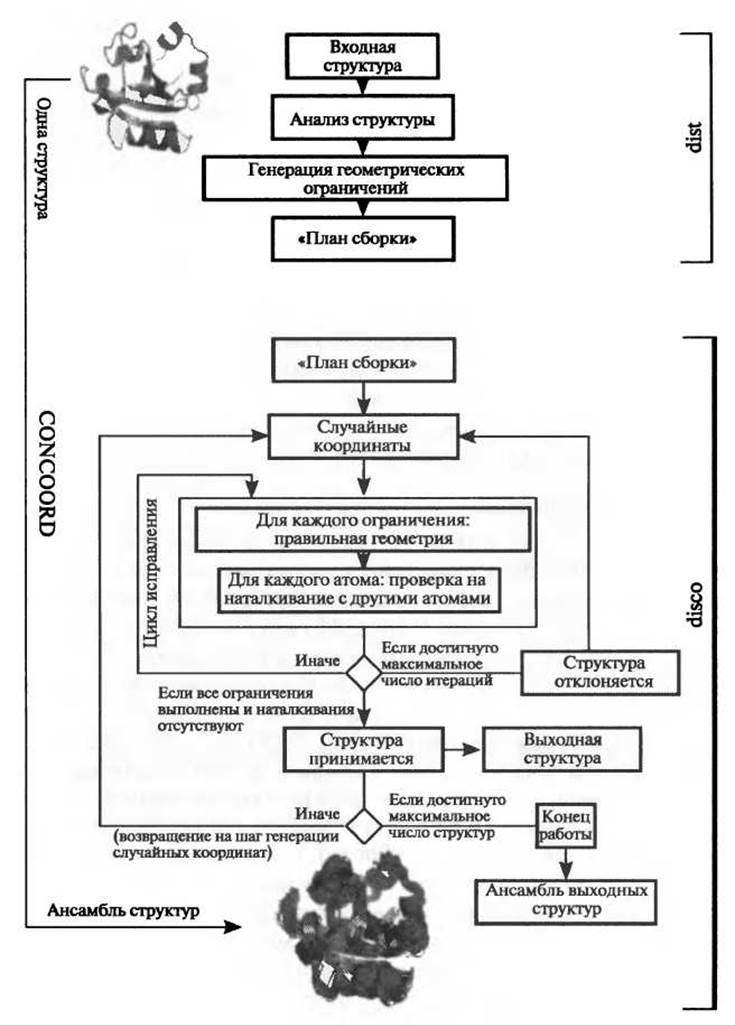
Рис. 9.11. Схематическое представление метода CONCOORD для создания структурных ансамблей из единственной исходной структуры. На первом шаге (программа dist) исходная структура анализируется и превращается в геометрическое описание белка. На втором шаге (программа disco), стартовав из случайных координат, структура воссоздается на основе определенных ранее ограничений
9.4.3.1. Приложения
CONCOORD и его недавно разработанное расширение tCONCOORD (Seeliger et al. 2007) были применены к рассмотрению различных белков. Аденилаткиназа демонстрирует отчетливое закрывающее движение доменов при связывании субстрата (АТФ/АМФ) или ингибитора (см. Рис. 9.13 вверху), причем СКО по Ca-атомам между конформацией со связанным лигандом и без него составляет 7.6 Å. С использованием программы tCONCOORD было выполнено два расчета, исходной конформацией для которых была выбрана закрытая (PDB код 1 АКБ). В одном из расчетов лиганд (Ар5А) был удален. На Рис. 9.13 внизу, показан результат анализа главных компонент для экспериментальных структур. Первый собственный вектор (ось абсцисс) соответствует домен-открывающему движению, показанному стрелкой на рис. 9.13 (внизу). Каждая точка на графике представляет одну структуру. Красные точки представляют ансамбль, который был получен исходя из закрытой конформации аденилаткиназы без лиганда. Зеленые точки представляют ансамбль, который был получен исходя из лиганд-связывающей конформации. Несмотря на то, что расчет с ингибитором позволил просэмплировать, главным образом, закрытые конформации в окрестности структуры с лигандом, расчет структуры без лиганда позволил просэмплировать как закрытую, так и открытую конформации, что дало возможность приблизиться к экспериментально полученным открытым конформациям с СКО 2.4, 2.6 и 3.1 Å для структур 1DVR, 1АК2 и 4АКЕ, соответственно. В создании лекарственных препаратов, которое опирается на знание структуры рецептора, часто возникает обратная задача - предсказание структуры рецептора с лигандом исходя из структуры свободного рецептора. Расчет, стартовавший из открытой конформации (4АКЕ), привел к получению структур, приближающихся к закрытой конформации с СКО 2.5, 2.9 и 3.3 Å для 1DVR, 1АК2 и 1АКЕ, соответственно. Таким образом, функциональное домен-открывающее движение было предсказано в обоих случаях: при использовании в качестве исходной как закрытой, лиганд-содержащей, конформации, так и открытой конформации, не содержащей лиганда.
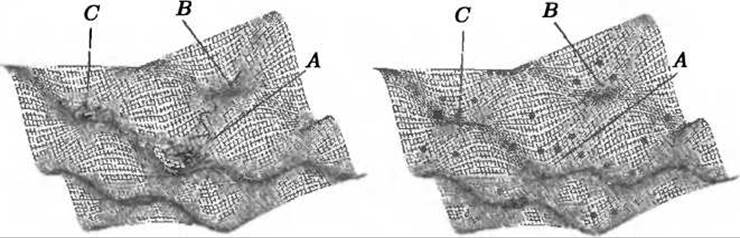
Рис. 9.12. Сравнение сэмплирующих свойств молекулярной динамики и метода CONCOORD на гипотетической энергетической поверхности. МД-траектория (слева) “гуляет” по поверхности, позволяя, таким образом, получить информацию о времени и пути между конформационными подсостояниями. CONCOORD (недетерминистически) “скачет” по поверхности, позволяя, таким образом, лучше просэмплировать конформационное пространство
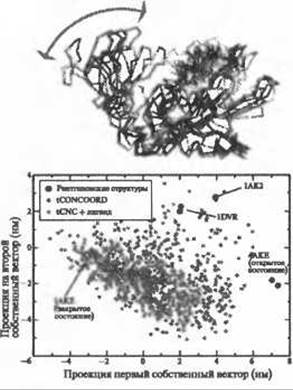
Рис. 9.13. (Цветную версию рисунка см. на вклейке.) Вверху: наложение рентгенографических структур аденилаткиназы. Внизу: анализ главных компонент. Два ансамбля структур, полученных по методу tCONCOORD, спроецированы на два первых собственных ветора, полученных при анализе главных компонент ансамбля рентгеновских структур. Ансамбль, представленный красными точками, построен исходя из закрытой конформации (1АКE) с удаленным ингибитором. Построенный ансамбль сэмплирует как закрытые, так и открытые конформации. Ансамбль, представленный зелеными точками, также построен исходя из закрытой конформации (1 АКБ), но с ингибитором. Этот ансамбль сэмплирует только закрытые конформации в окрестности исходной лиганд-содержащей
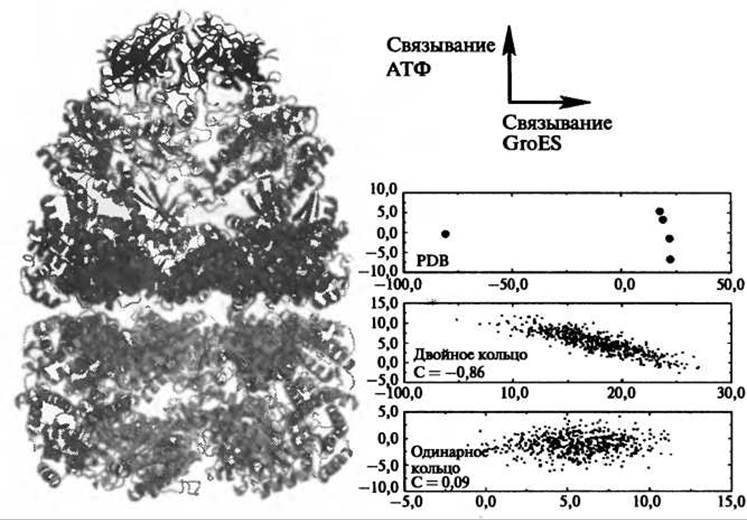
Рис. 9.14. Асимметричный комплекс GroEL-GroES (слева) вместе с результатами расчетов CONCOORD (справа). Комплекс GroEL-GroES состоит из двух кошаперонинов GroES (показаны черным), транс-кольца GroEL, связанного с GroES, (показано темно-серым) и цис-кольца (показано светло-серым). Анализ главных компонент выявил два главных структурных перехода для кольца GroEL: после связывания нуклеотида (вертикальная ось на правой панели) и после связывания GroES (горизонтальная ось), соответственно. В расчетах двойного кольца, но не одинарного, эти моды оказались связанными, предполагая наличие связи между внутрикольцевой и межкольцевой кооперативностью
Благодаря своей вычислительной эффективности CONCOORD может быть широко использован для выявления функционально значимых мод подвижности для таких молекулярных систем, которые лежат за пределами ограничений по размеру, свойственными другим методикам расчетов на атомарном уровне, например, молекулярной динамике. Применение CONCOORD к комплексу шаперонинов GroEL-GroES, содержащему более 8 000 остатков, выявило новую форму связи между внутрикольцевой и межкольцевой корпоративностью (de Groot et al. 1999). Каждое кольцо GroEL продемонстрировало две основных моды коллективного движения: основной конформационный переход при связывании с кошаперонином GroES и вторичный переход при связывании АТФ (Рис. 9.14 вверху справа). Расчет с помощью CONCOORD одного лишь кольца GroEL не показал какой-либо связи между этими модами, в то время как расчет системы двух колец оказал четкую корреляцию между двумя этими модами, объясняя, таким образом, как связывание нуклеотидов влияет на аффинность к Gro ES в двойном кольце, но не в одинарном.