Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Интегральные серверы для предсказания функции по структуре
ProFunc
Вторым интегральным сервером, описываемым тут, является ProFunc (Laskowski et al. 2005b) (http://www.ebi.ac.uk/profunc), созданный в Европейском институте биоинформатики (EBI) в рамках сотрудничества с Центром по структурной геномике на Среднем Западе (Midwest Center for Structural Genomics, MCSG). Сервер ProFunc позволяет пользователю либо самому загрузить структуру белка, либо ввести PDB-код структуры, уже имеющейся в базе данных белковых структур. В последнем случае, если сервер однажды уже выполнял расчет для этой структуры, то результат появится незамедлительно. Когда ProFunc выполняет расчет, то, как показано на рисунке 10.4, он использует ряд методов, основанных на последовательности белка и его структуре, причем произволится такое распараллеливание расчетов, что разные методы выполняются на разных процессорах. Некоторые из ресурсоемких методов сами тоже запускаются параллельно на множестве процессоров. Весь расчет заканчивается обычно в течение часа.
Затем результаты всех методов суммируются, причем детали результатов каждого метода остаются доступными. Однако здесь результаты не объединяются таким изощренным образом, как это делается в ProKnow. Вместо этого наверху страницы с результатами приводится краткий итог, показывающий наиболее общие термы по системе ГО и названия белков, но это следует рассматривать лишь как краткую инструкцию. Основная цель сервера состоит в представлении результатов в легкодоступном виде, чтобы дать исследователям возможность интерпретировать их, используя свой собственный опыт и информацию о рассматриваемом белке.
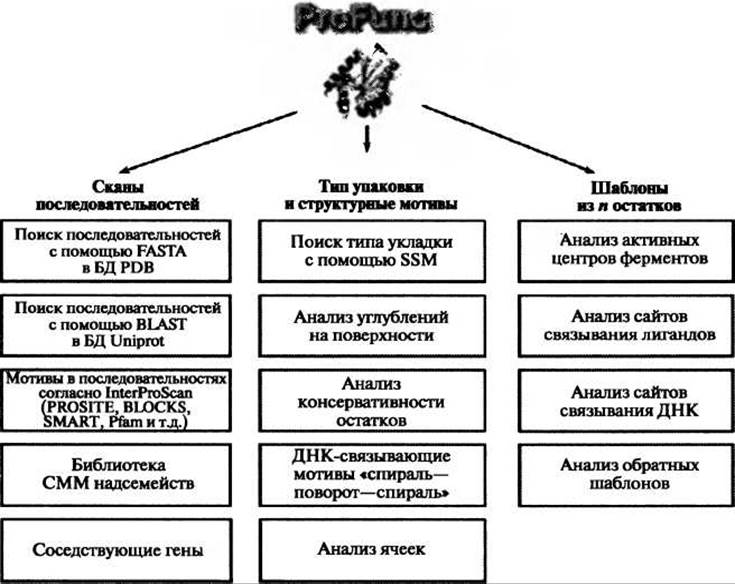
Рис. 10.4. Схематическая диаграмма используемых в ProFunc методов, основанных на последовательности и структуре. Методы анализа последовательности, перечисленные в левом столбце, включают поиск последовательности белка в базах PDB и Uniprot. Поиск с помощью InterProScan и Superfamily выявляет любые мотивы в последовательностях из соответствующих им баз данных, которые есть в последовательности рассматриваемого белка. Для каждого подходящего варианта из Uniprot, отобранного программой BLAST, по возможности выполняется локализация соответствующего ему гена в геноме и определение всех соседних генов. В среднем столбце первый из методов поиска, основанных на структуре, использует программу SSM для определения структур, общая укладка которых наиболее сходна с укладкой рассматриваемого белка. Затем рассчитываются углубления на поверхности, которые могут быть визуализованы с раскраской по типу образующих эти углубления остатков или их консервативности. Затем определяется два типа структурных мотивов: мотивы “спираль-поворот-спираль” (helix-tum-helix, НТН), характерные для многих ДНК- связывающих белков, и ячейки, которые часто обнаруживаются в функционально важных местах. Наконец, в правом столбце представлены различные методы шаблонов для поиска локальных пространственных совпадений с известными структурами белков
Теперь, хотя ProFimc и использует ряд методов, основанных на последовательности, включая хорошо известные методы, такие как FASTA и InterProScan (Quevillon et al. 2005), мы опишем только методы, основанные на структуре, поскольку большинство из них уникальны.