Структура и функционирование белков. Применение методов биоинформатики - Джон Ригден 2014
Интегральные серверы для предсказания функции по структуре
ProFunc
Основанные на структуре методы, используемые ProFunc
10.3.1.1. Поиск совпадений типа укладки
Первым из методов, основанных на структуре, является поиск в представительной выборке из базы данных PDB структур с таким же, или схожим, типом укладки, что и у рассматриваемой структуры. Для этого используется программа SSM (Secondary Structure Matching, Сопоставление вторичной структуры) (Krissinel and Henrick 2004). Эта программа производит быструю процедуру сопоставления графов для сравнения элементов вторичной структуры рассматриваемой структуры с такими элементами структур в базе данных. Хорошо совпавшие структуры накладываются й для них рассчитывается СКО по эквивалентным Са-атомам, а также стандартизованная мера значимости и собственная мера значимости SSM, называемая Q-показатель. ProFunc показывает десять лучших совпадений, упорядоченных по Q-показателю, и любое из них, или все сразу, могут быть наложены на рассматриваемую структуру при помощи программы молекулярной графики RasMol (Sayle and Milner-White 1995).
Лучшее совпадение типа укладки для нашего примера, структуры 2fck, показано на рисунке 10.5. Этим совпадением является структура из PDB с кодом ls7f, представляющая собой RimL N(а)-ацетилтрансферазу из Salmonella typhimurium (Vetting et al. 2005). Этот белок отвечает за превращении прокариотического рибосомального белка из L12 в L7 путем ацетилирования его N-концевой аминогруппы. Белок образует гомодимер и интерфейс димера образует большой желоб, способный к связыванию N-концевой спирали L12. На некотором удалении от сайта связывания субстрата белок связывает также кофермент А (Ко А).
10.3.1.2. ДНК-связывающий мотив “спираль-поворот-спираль”
Вторым основанным на структуре методом является поиск каких-либо мотивов “спираль-поворот-спираль” (helix-tum-helix, НТН), которые совпадают с мотивами, извлеченными из участвующих в связывании ДНК структур PDB (Jones et al. 2003; Aravind et al. 2005). Ложноположительные результаты отсеиваются по ряду параметров, основанных на сочетании доступности растворителю и электростатического потенциала (Shanahan et al. 2004), поэтому любой положительный результат может указывать, что рассматриваемый белок связывает ДНК, хотя, конечно, это сообщает мало нового о функции белка. В нашем примере структура 2fck не имеет мотивов НТН, как мы и ожидали.
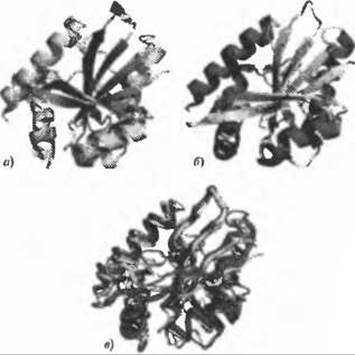
Рис. 10.5. (Цветную версию рисунка см. на вклейке.) Наиболее близкий к структуре 2fck тип укладки, обнаруженный с помощью программы поиска совпадающих типов укладки SSM в структуре 1s7f, RimL NІ(а)-ацегилтраснферазе из Salmonella typhimurium. а) Общая пространственная структура 2fck и б) общая структура 1s7f в той же ориентации, в) Структуры совмещены и показаны в виде линии Са-атомов - 2fck желтым, a 1s7f фиолетовым. Совпадающие области выделены более толстыми линиями в каждой структуре
10.3.1.3. Ячейки
Третий метод выявляет в структуре мотивы типа “ячейка”, которые часто связаны с функциональными сайтами. Ячейка - это сайт связывания аниона или катиона, образованный тремя или более аминокислотными остатками, у которых двугранные углы основной цепи (ψ-(р) изменяются между а- и у-областями на карте Рамачандрана, соответствующими право- и левозакрученным спиралям (Watson and Milner-White 2002а, b). Как и прежде, визуализация в программе RasMol показывает расположение ячеек в контексте всей пространственной структуры. Сервер ProFunc присваивает каждой ячейке оценку, исходя из следующих критериев: количество доступных растворителю атомов NH, консервативность составляющих ячейку остатков и того, находится ли ячейка в более крупном углублении поверхности. Ячейки могут оказаться полезными, когда ни один из других методов ничего не может сказать о функции белка. В таких случаях расположение ячеек может указать на предположительно функционально важные места в пространственной структуре белка.
Структура 2fck содержит несколько таких ячеек, три из которых имеют достаточно высокую оценку, что говорит об их потенциальной функциональной значимости. И в самом деле, ячейка, имеющая наиболее высокую оценку, расположена в вероятном сайте связывания субстрата (по аналогии со сходной структурой ls7f), в то время как вторая и третья ячейки обнаружены возле ворот сайта связывания соА.
10.3.1.4. Углубления на поверхности
Затем с помощью программы SURFNET (Laskowski 1995) рассчитываются все углубления на поверхности белка. Углубления ранжируются по размеру и могут быть визуализованы с помощью RasMol. Опции визуализации позволяют раскрасить углубления по их специфическим свойствам: размеру углубления, типу остатков или их консервативности. Размер важен, поскольку самое большое углубление на поверхности белка обычно находится в месте расположения его активного центра (Laskowski et al. 1996). Консервативность остатков также важна, поскольку группа высококонсервативных остатков, особенно расположенная в большом кармане, с большой вероятностью указывает на функциональный сайт (Lichtarge and Sowa 2002; Madabushi et al. 2002; Glaser et al. 2003). Как и анализ ячеек, изучение углублений приносит наибольшую пользу, когда остальные методы потерпели неудачу или предлагают только неопределенные варианты. В нашем случае наибольшее углубление действительно соответствует предполагаемому сайту связывания белка, совпадающему по расположению со связанным коА в родственных структурах, выявленных по совпадению типа укладки методами, рассмотренными выше, и методами шаблонов, которые описаны ниже.
10.3.1.5. Методы шаблонов
Последние методы, используемые сервером ProFunc, включают в себя четыре различных типа поиска по шаблону из остатков (Laskowski et al. 2005с). По определению, шаблоны - это особые пространственные конформации, как правило, трех аминокислотных остатков. Поиск по шаблонам выполняется быстрым алгоритмом пространственного поиска, называемым JESS (Barker and Thornton 2003), который запускается параллельно на нескольких процессорах.
Шаблоны ферментов
Первую группу шаблонов составляют шаблоны активных центров ферментов, которые взяты из составленного вручную Атласа каталитических центров (Catalytic Site Atlas, CSA) (Porter et al. 2004). Здесь каждый шаблон имеет от двух до пяти остатков, которые либо описаны в литературе, как каталитические, либо являются высококонсервативными и лежат в непосредственной близости к каталитическим остаткам. Хорошее совпадение (см. ниже) с одним из таких шаблонов может оказаться явным указанием на функцию белка.
Шаблоны связывания лигандов и ДНК
Две следующие группы шаблонов - это шаблоны связывания лигандов и ДНК, которые автоматически генерируются раз в неделю, чтобы учитывать все обновления, произошедшие в базе данных PDB. Шаблоны связывания лигандов генерируются поочередным рассмотрением каждого типа гетерогрупп (согласно словарю гетерогрупп (Het Group Dictionary) в PDB) и составлением списка негомологичных PDB-структур, содержащих эти гетерогруппы. Сначала отмечаются остатки, взаимодействующие с гетерогруппами в каждой из выбранных структур. Затем определяются шаблоны, состоящие из групп по три остатка из ранее отмеченных, и сохраняются как шаблоны для соответствующих гетерогрупп. Есть следующие критерии отбора, определяющие, какая тройка остатков может считаться шаблоном: каждый остаток должен находиться не далее 5 Å от других остатков шаблона, каждый шаблон может иметь не более одного гидрофобного остатка (т.е. Ala, Phe, Ile, Leu, Met, Pro или Val), - это необходимо, чтобы настроить шаблоны на остатки поверхности, - и не должно быть двух шаблонов из одной структуры, имеющих более одного общего остатка. Порядок рассмотрения потенциальных шаблонов определяется их относительной значимостью. Так, шаблон, содержащий остатки, образующие несколько водородных связей с данной гетерогруппой, более значим, чем те шаблоны, в которых остатки лишь слегка взаимодействуют с гетерогруппой. Шаблоны связывания ДНК генерируются точно таким же образом, за исключением того, что все молекулы ДНК и РНК рассматриваются как единая гетерогруппа. По состоянию на май 2008 года в этой базе данных насчитывалось 584 шаблона каталитических центров, 97,534 шаблонов связывания лигандов и 3,390 шаблонов связывания ДНК. На рисунке 10.6 показано совпадение шаблона в структуре 2fck с шаблоном связывания коА в структуре ls7n, RimL N(а)-ацетилтрансферазе из Salmonella typhimurium.
Обратные шаблоны
Четвертая группа шаблонов посвящена поиску любых совпадений, которые могли ускользнуть от первых трех групп. Это обратные шаблоны, которые вычисляются по самой рассматриваемой структуре. Они генерируются с использованием в основном тех же самых правил, что шаблоны связывания лигандов и ДНК. Главное различие состоит в том, что, во- первых, рассматривается вся структура белка целиком, а не только остатки, контактирующие с лигандами или ДНК, и, во-вторых, каждому шаблону присваивается вес в зависимости от консервативности входящих в него остатков (которая рассчитывается по сделанному с помощью программы BLAST множественному выравниванию последовательностей, взятых из базы данных последовательностей UniProt). Шаблоны выбираются таким образом, что, в идеале, каждый остаток белка представлен по крайней мере в одном шаблоне, хотя если шаблонов получается слишком много, то их число сокращается до удвоенного числа остатков в последовательности.
Лучший обратный шаблон к структуре 2fck показан на рис. 10.7. Совпадением является структура ls7f, RimL N(а)-ацетилтрансфераза из S. typhimurium. Это апо-форма структуры ls7n, совпадение с которой было выявлено с помощью вышеописанных программы поиска совпадений вторичной структуры SSM и шаблона связывания лигандов.
Поиск по шаблонам и их оценка
Поиск по шаблону может дать сотни, тысячи и даже десятки тысяч совпадений, особенно в случае обратных шаблонов. Поэтому задача состоит в том, чтобы отбросить случайные совпадения и оставить только значимые, отсортировав их по значимости. ProFunc делает это, сравнивая окружение остатков шаблона в его родительской структуре с окружением совпадающих с ним остатков в структуре рассматриваемой. Остаткам родительской структуры в радиусе 10 Å от геометрического центра шаблона согласно степени сходства и перекрывания ставятся в соответствие остатки из такой же области рассматриваемой структуры. В случаях альтернативных вариантов создания таких пар применяется процедура оптимизации, направленная на максимизацию числа пар идентичных или схожих остатков с эквивалентным положением в пространстве. Число пар остатков дает грубую оценку локального сходства совпадающих сайтов в двух белках (Рис. 10.6b и 10.7b). Однако и эта грубая оценка все еще оставляет слишком много ложно-положительных совпадений. Поэтому применяемая в действительности оценка учитывает относительное расположение составляющих пару остатков в соответствующих аминокислотных последовательностях. Если составляющие пару остатки следуют в одинаковом порядке в обоих последовательностях, тогда высока вероятность гомологичности последовательностей.
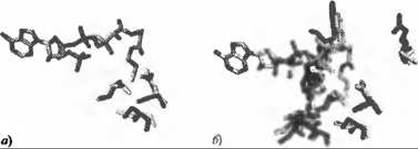
Рис. 10.6. (Цветную версию рисунка см. на вклейке.) Совпадение шаблонов связывания лиганда по данным сервера ProFunc. а) Три остатка, показанных фиолетовым, соответствуют шаблону связывания лиганда для лиганда из кофермента А (коА), показанному раскраской по типу атомов (углерод серый, азот синий, кислород красный, сера желтая и фосфор оранжевый). Шаблон образован остатками Asn138, Ser141 и Cys134 из PDB-структуры 1s7n, RimL N(а)-ацетилтрансферазы из Salmonella typhimurium. Три остатка, показанные желтым, - это остатки из рассматриваемой структуры 2fck, совпадающие с остатками шаблона. Это Asn140, Ser143 аи Cys136, соответственно. CKO по 14 атомам боковых цепей составляет 1.18 Å. б) Как и на рисунке (а), но дополнительно показаны совпадающие остатки, лежащие в радиусе 10 А от центра шаблона. Это остатки того же типа, которые совмещаются при наложении рассматриваемой структуры и шаблонной структуры. Фиолетовым показаны остатки из шаблонной структуры (1s7n), желтым - из рассматриваемой структуры (2fck)
Чтобы увидеть, почему это так, рассмотрим две имеющие общего предка последовательности, которые разошлись настолько далеко, что их родство не может быть установлено методами анализа последовательностей. Однако если обе они сохранили одну и ту же функцию, то областью, которая претерпела наименьшие изменения, вероятно будет активный центр, поскольку любое изменение в нем изменило бы и функцию. Сухой остаток этого рассуждения состоит в том, что самый высокий уровень сходства между двумя белками будет среди остатков в окрестности активного центра. Эти остатки будут близки в пространстве, но могут быть разбросаны по последовательности этих белков. Вот почему можно обнаружить сходство в пространственной структуре, но практически невозможно уловить его при сравнении последовательностей.
Иллюстрация этому приведена на рисунке 10.7с, где представлено выравнивание последовательностей между структурой 2fck и наиболее сходной с ней по обратному шаблону структурой ls7f. Выравнивание задавалось остатками, которые были определены как эквивалентные в процедуре поиска локального совпадения, описанной выше. Эти остатки отмечены двумя точками между последовательностями. (Одна точка соответствует остаткам, которые утратили своих пространственно-эквивалентных партнеров в выравнивании). Легко видеть, что составляющие пары остатки, лежащие в компактной области пространства, распределены почти по всей длине обеих последовательностей.
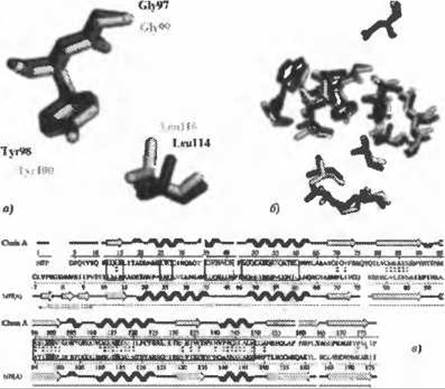
Рис. 10.7. (Цветную версию рисунка см. на вклейке.) Совпадение обратных шаблонов между структурами 2fck и 1s7f, соответствующими RimL N(а)-ацетилтрансферазе из Salmonella typhimurium. а) Желтым показаны остатки шаблона из 2fck (Gly99, Туr100 и Leu 116), которые соответствуют остаткам из 1s7f (Gly97, Туr98 и Leu114, соответственно), показанным фиолетовым, с СКО 0.62 Å по 17 совпадающим атомам, б) Эквивалентные остатки одинаковых типов в радиусе 10 Å от центра шаблона. 16 таких остатков (из всего 44 на удалении 10 Å и менее) дают 36.4% локальной идентичности. Следующие 20 остатков (не показаны) имеют сходный тип (например, не соответствует Val), в) Выравнивание структур, полученное из наложения структур. Верхняя строка соответствует вторичной структуре в 2fck, а нижняя - в 1s7f; спирали показаны зубчатыми элементами, ß-тяжи стрелками. Три выделенных остатка в выравнивании последовательностей соответствуют остаткам шаблона. Двойные точки между последовательностями обозначают остатки, содержащиеся внутри сферы с радиусом 10 Å и центром в центре шаблона, т.е. те остатки, которые были использованы для выполнения выравнивания. Области в рамках представляют сегменты выравнивания, где идентичность последовательностей превышает 35%. Длинные тонкие стрелки снизу показывают структурно подходящие области, т.е. сегменты обоих белков, где Ca-атомы могут быть совмещены по структуре с СКО менее 3.0 Å
Еще более интересно, что в то время как выравнивание в целом дает 24,7% идентичности последовательностей двух белков, идентичными оказываются 16 из 44 остатков в радиусе 10 Å от центра шаблона, что дает локальную идентичность 36,4%. Поскольку эта область соответствует значительной части сайта связывания коА в структуре ls7f, это является сильным структурным аргументом в пользу того, то структура 2fck также связывает коА. Область также захватывает и часть предполагаемого сайта связывания субстрата, но её недостаточно, ни чтобы сделать вывод об одинаковом субстрате для обоих белков, ни чтобы предположить у них одинаковую функцию.
В дополнение к оценке локального сходства, ProFunc использует и другие статистические показатели. Одним из них является математическое ожидание Е (E-value), связанное с этой оценкой. Для обратных шаблонов Е вычисляется из распределения всех оценок, полученных в данном поиске, по той же процедуре, что используется в программе FASTA (Pearson 1998). При поиске по другим шаблонам математическое ожидание Е вычисляется с использованием заранее рассчитанных параметров. Лучшие результаты сортируются по значению Е на четыре группы: достоверное совпадение (Е < 10-6), весьма вероятное совпадение (10-6 < Е < 0.01), вполне вероятное совпадение (0.01 < Е < 0.1) и маловероятное совпадение (0.1 < Е < 10.0).
Также используется общее структурное сходство и наибольшая протяженность фрагментов последовательностей, которые еще можно наложить с СКО 3.0 Å по Ca-атомам. Последний показатель может быть полезным, когда есть длинное перекрывание, предполагающее значительное структурное совпадение даже в случае маловероятных совпадений.
10.3.1.6. Структурный анализ с помощью PDBsum
Не очень существенным для предсказания функции, но полезным побочным эффектом загрузки структуры на сервер ProFunc является создание для этой структуры нескольких страниц в атласе PDBsum. Это красочно иллюстрированный атлас белковых структур (http://www.ebi.ac.uk/pdbsum), выполняющий ряд анализов структуры для загруженных белков и представляющий результаты с помощью различных схематических диаграмм (Laskowski et al. 2005а). Пара примеров приведена на рис. 10.8.