БІОХІМІЯ - Підручник - Остапченко Л. І. - 2012
Розділ 6. ОБМІН І ФУНКЦІЇ АМІНОКИСЛОТ. БІОСИНТЕЗ БІЛКА
6.8. Обмін окремих амінокислот
6.8.3. Обмін фенілаланіну й тирозину
Фенілаланін - незамінна амінокислота, тому що в клітинах тварин не синтезується її бензольне кільце. Тирозин - умовно замінна амінокислота, оскільки утворюється з фенілаланіну. Вміст цих амінокислот у харчових білках, у тому числі й рослинних, досить невеликий. Фенілаланін і тирозин використовуються для синтезу багатьох біологічно активних сполук. У різних тканинах метаболізм цих амінокислот відбувається по-різному (рис. 6.28).
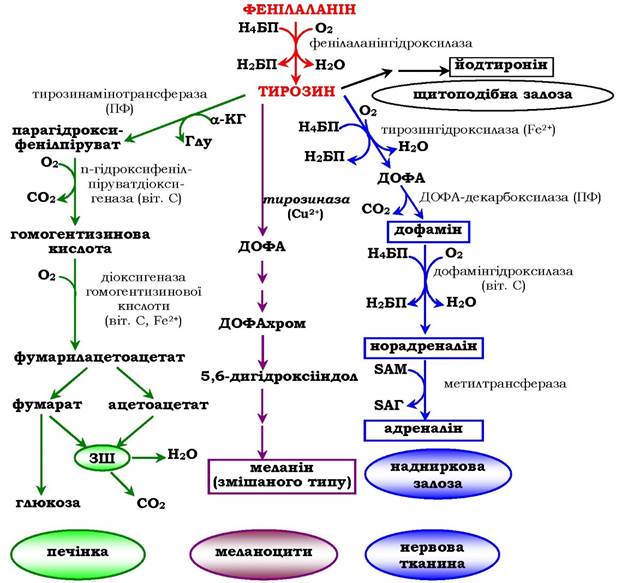
Рис. 6.28. Шляхи перетворення фенілаланіну і тирозину в різних тканинах:
Н4БП - тетрагідробіоптерин; Н2БП - дигідробіоптерин;
ПФ - піридоксальфосфат; SAM - S-аденозилметіонін
Основна кількість фенілаланіну використовується двома шляхами:
✵ включається в білки;
✵ перетворюється на тирозин.
Перетворення фенілаланіну на тирозин передусім необхідне для видалення надлишку фенілаланіну, осуільки його високі концентрації токсичні для клітин. Утворення тирозину не має великого значення, адже нестачі цієї амінокислоти в клітинах практично не спостерігається.
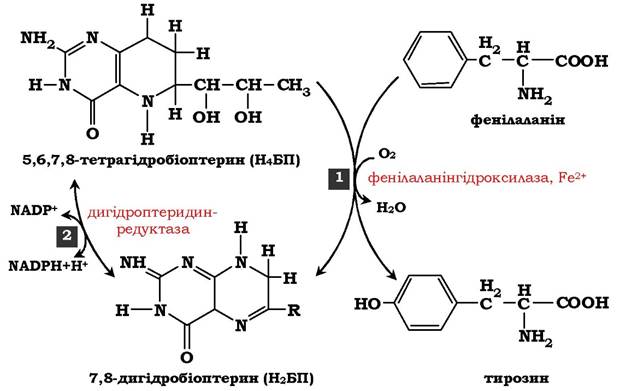
Основний шлях метаболізму фенілаланіну починається з його гідроксилювання (рис. 6.29), у результаті чого утворюється тирозин. Ця реакція каталізується специфічною монооксигеназою - фенілаланінгідроксилазою, коферментом якої слугує тетрагідробіоптерин (Н4БП). Активність ферменту залежить також від наявності Fe2+. Реакція є необоротною: Н4БП у результаті реакції окиснюється до дигідробіоптерину (Н2БП). Регенерація останнього відбувається за участю дигідроптеридинредуктази з використанням НАДФН + H+.
Обмін тирозину набагато складніший, ніж обмін фенілаланіну. Крім використання в синтезі білків, тирозин у різних тканинах виступає попередником таких сполук, як катехоламіни, тироксин, меланіни, і катаболізується до СО2 і Н2О.
У печінці катаболізм тирозину відбувається до кінцевих продуктів. Специфічний шлях катаболізму включає декілька ферментативних реакцій і завершується утворенням фумарату й ацетоацетату:
1) Трансамінування тирозину з α-кетоглутаратом каталізує тирозин-амінотрансфераза (коферменнт ПФ) - індукований фермент печінки ссавців. У результаті проходження реакції утворюється п-гідроксифенілпіруват.
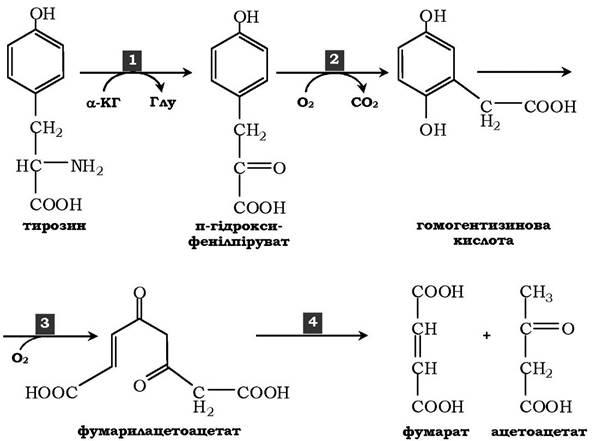
2) У реакції окиснення п-гідроксифенілпірувату до гомогентизинової кислоти відбувається декарбоксилювання, гідроксилювання ароматичного кільця й міграція бічного ланцюга. Реакцію каталізує фермент п-гідроксифенілпіруватдіоксигеназа, кофакторами якого є вітамін С і Fe2 +.
3) Перетворення гомогентизинової кислоти на фумарилацетоацетат супроводжується розщепленням ароматичного кільця. Ця реакція каталізується діоксигеназою гомогентизинової кислоти, що містить Fe2+ як кофермент.
Обмін фенілаланіну й тирозину пов'язаний зі значною кількістю реакцій гідрокислювання, які каталізують оксигенази. Ферменти оксигенази (гідроксилази) використовують молекулу О2 і кофермент-донор водню (найчастіше Н4БП). Для каталізу оксигеназам необхідні кофактори - Fe2+ або гем (для деяких - Cu+), а для багатьох ще й вітамін С. Оксигенази поділяють на дві групи:
✵ монооксигенази - приєднують один атом з О2 до продукту реакції, другий використовують для утворення Н2О;
✵ діоксигенази - використовують обидва атома О2 для утворення продукту реакції.
Практично всі процеси розщеплення ароматичних кілець у біологічних системах каталізуються діоксигеназами — підкласом ферментів, який відкрив японський біохімік Осамі Хайяші. У результаті розривання бензольного кільця утворюється малеїлацетоацетат, який в процесі цис- і транс-ізомеризації перетворюється на фумарилацеоацетат.
4) Гідроліз фумарилацетоацетату під дією фумарилацетоацетатгідролази зумовлює утворення фумарату й ацетоацетату. Фумарат може окиснюватися до СО2 і Н2О або використовуватися для глюконеогенезу. Ацетоацетат - кетонове тіло, що окиснюється до кінцевих продуктів з виділенням енергії.
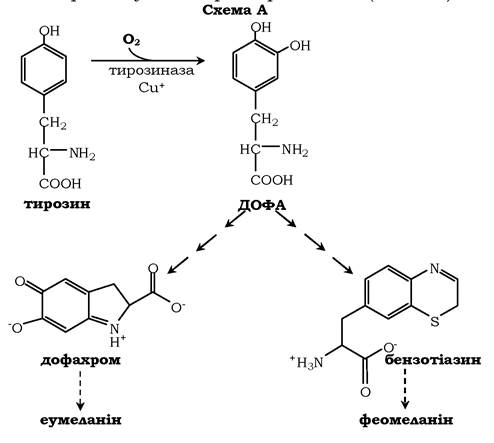
У пігментних клітинах (меланоцитах) тирозин є попередником темних пігментів - меланінів. Серед них переважають два типи: еумеланіни і феомеланіни. Еумеланіни (чорного й коричневого кольору) - нерозчинні високомолекулярні гетерополімери 5,6-дигідроксііндолу й деяких його попередників. Феомеланіни - жовті або червонувато-коричневі полімери, розчинні в розведених лугах. Містяться вони в основному у волоссі. Меланіни присутні й в сітківці ока. Колір шкіри залежить від розподілу меланоцитів і кількості в них різних типів меланінів.
Синтез меланінів - це складний, багатоступеневий, розгалужений процес (коротку схему синтезу див. на рис. 6.28). Першу реакцію - перетворення тирозину на ДОФА - каталізує тирозиназа, яка використовує як кофактор іони Cu+ (схема А):
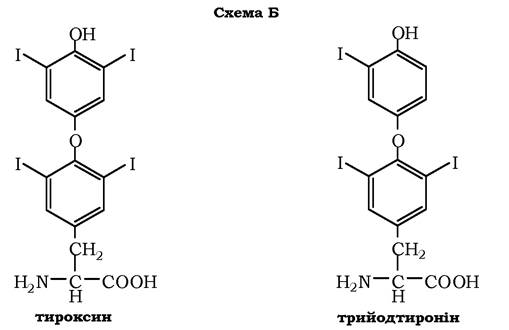
У щитоподібній залозі синтезуються й виділяються гормони йодтироніни: тироксин (тетрайодтиронін) і трийодтиронін - йодовані залишки тирозину, котрі потрапляють до клітин щитоподібної залози через базальну мембрану (схема Б):
У мозковій речовині надниркових залоз і нервовій тканині тирозин є попередником катехоламінів (дофаміну, норадреналіну та адреналіну):
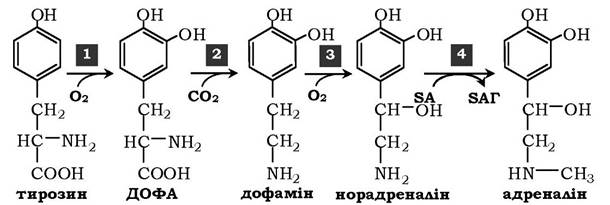
Під час утворення катехоламінів, яке відбувається в нервовій тканині й надниркових залозах, і меланіну в меланоцитах проміжним продуктом слугує діоксифенілаланін (ДОФА). Але гідроксилювання тирозину в клітинах різних типів каталізується різними ферментами:
✵ тирозиназа в меланоцитах є Сu+-залежним ферментом;
✵ тирозингідроксилаза (1) у надниркових залозах і катехоламінергічних нейронах не потребує іонів міді. Це - Fе2+-залежний фермент, що аналогічно до фенілаланінгідроксилази як кофермент використовує Н4БП. Фізіологічна роль тирозингідроксилази надзвичайно велика, оскільки цей фермент є регуляторним і визначає швидкість синтезу катехоламінів. Активність тирозингідроксилази значно змінюється унаслідок, по-перше, алостеричної регуляції (інгібітор - норадреналін); по-друге, фосфорилювання/дефосфорилювання: у результаті фосфорилювання за участю протеїнкінази А знижуються Km для коферменту Н4БП і спорідненість ферменту до норадреналіну, унаслідок чого відбувається активація тирозингідроксилази. Кількість ферменту регулюється на рівні транскрипції;
✵ ДОФА-декарбоксилаза (2) (кофермент - ПФ) каталізує утворення дофаміну, який за участю дофамінгідроксилази (3) (монооксигенази) перетворюється в норадреналін. Для функціонування ферменту необхідні іони Cu+, вітамін С і тетрагідробіоптерин;
✵ у мозковій речовині надниркових залоз фенілетаноламін-N-метилтрансфераза (4) каталізує метилювання норадреналіну, у результаті утворюється адреналін. Джерелом метильної групи слугує SAM.
Дофамін і норадреналін слугують медіаторами в синаптичній передачі нервових імпульсів, а адреналін є гормоном широкого спектра дії, який регулює енергетичний обмін. Одна з функцій катехоламінів - регуляція діяльності серцево-судинної системи.
Відомо кілька спадкових хвороб, пов'язаних з дефектом ферментів обміну фенілаланіну й тирозину в різних тканинах. У печінці здорових людей невелика частина фенілаланіну (~10 %) перетворюється на феніллактат і фенілацетилглутамін (рис. 6.30).
Цей шлях катаболізму фенілаланіну стає головним під час порушення основного шляху - перетворення на тирозин, який каталізується фенілаланінгідроксилазою. Таке порушення супроводжується гіперфенілаланінемією й підвищенням у крові та сечі вмісту метаболітів альтернативного шляху: фенілпірувату, фенілацетату, феніллактату й фенілацетилглутаміну. Дефект феніла ланінгідрокслази є причиною такого захворювання, як фенілкетонурія (ФКУ). Існують дві її форми:
✵ класична ФКУ - спадкове захворювання, пов'язане з мутаціями в гені фенілаланінгідроксилази, які приводять до зниження активності ферменту або повної його інактивації. При цьому концентрація фенілаланіну в крові підвищується у 20-30 разів (у нормі - 1,0-2,0 мг/дл), у сечі - у 100-300 разів порівняно з нормою (30 мг/дл). Концентрація фенілпірувату й феніллактату в сечі досягає 300-600 мг/дл, тоді як у нормі повністю відсутні.
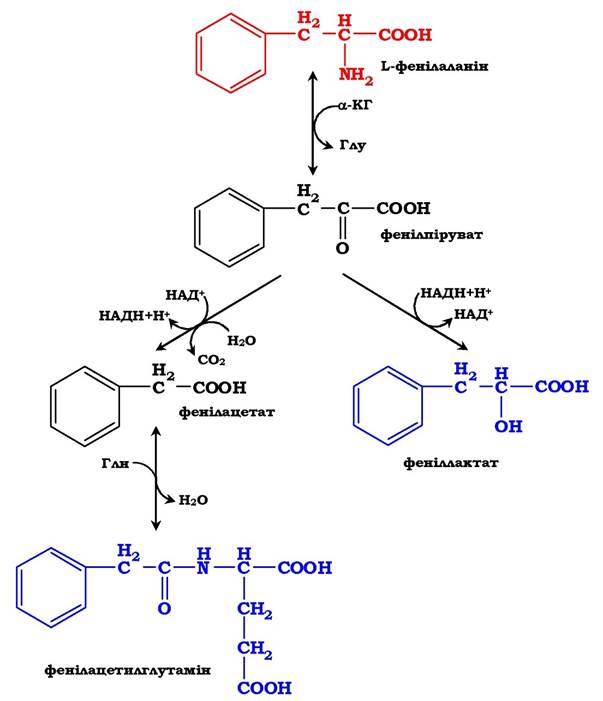
Рис. 6.30. Альтернативні шляхи катаболізму фенілаланіну.
Унаслідок дефекту фенілаланінгідроксилази накопичений фенілаланін піддається трансамінуванню з α-кетоглутаратом. Утворений фенілпіруват перетворюється або на феніллактат, або на фенілацетилглутамін, які накопичуються в крові та виділяються з сечею.
Ці сполуки токсичні для клітин мозку.
Найважчі прояви ФКУ - порушення розумового, фізичного розвитку та пігментації, судомний синдром. У разі відсутності лікування хворі не доживають до 30 років. Частота захворювання становить 1 : 10000. Спадкується за аутосомно-рецесивним типом.
Важкі прояви ФКУ пов'язані з токсичною дією на клітини мозку високих концентрацій фенілаланіну, фенілпірувату, феніллактату. Великі концентрації фенілаланіну обмежують транспорт тирозину та триптофану через гематоенцефалічний бар'єр і гальмують синтез нейромедіаторів (дофаміну, норадреналіну, серотоніну).
✵ варіантна ФКУ (коферментзалежна гіперфенілаланінемія) виникає як наслідок мутацій у генах, котрі контролюють метаболізм Н4БП. Клінічні прояви майже збігаються з проявами класичної ФКУ. Частота захворювання - 1-2 випадки на 1 млн новонароджених.
Н4БП необхідний для реакцій гідроксилювання не тільки фенілаланіну, але й тирозину та триптофану, тому в разі нестачі цього коферменту порушується метаболізм усіх трьох амінокислот, у тому числі й синтез нейромедіаторів. Захворювання характеризується важкими неврологічними порушеннями та ранньою смертю ("злоякісна ФКУ»).
Прогресуюче порушення розумового та фізичного розвитку в дітей, хворих на ФКУ, можна попередити дієтою з дуже низьким вмістом або повним виключенням фенілаланіну. Якщо таке лікування почати відразу після народження дитини, то пошкодженню мозку можна запобігти. Вважається, що обмеження в харчуванні можуть бути послаблені після 10-річного віку (закінчення процесів мієлінізації мозку), але багато сучасних педіатрів схиляються в бік "пожиттєвої дієти".
Для діагностики ФКУ використовують якісні й кількісні методи виявлення паталогічних метаболітів у сечі, визначення концентрації фенілаланіну в крові й сечі. Дефектний ген, відповідальний за фенілкетонурію, можна виявити у фенотипічно нормальних гетерозиготних носіїв за допомогою тесту толерантності до фенілаланіну. Для цього обстежуваному дають натщесерце ~10 г фенілаланіну у вигляді розчину, потім через годинні інтервали беруть проби крові, в яких визначають вміст тирозину. У нормі концентрація тирозину в крові після фенілаланінового навантаження є значно вищою, ніж у гетерозиготних носіїв гену фенілкетонурії. Цей тест використовується в генетичній консультації для визначення ризику народження хворої дитини. Для виявлення новонароджених дітей з ФКУ розроблено схему скринінгу. Чутливість тесту досягає практично 100 %.
У наш час діагностику мутантного гену, відповідального за ФКУ, можна проводити за допомогою методів ДНК-діагностики (рестрикційного аналізу і ПЛР).
Деякі порушення катаболізму тирозину в печінці приводять до тирозинемії та тирозинурії. Розрізняють три типи тирозинемії.
Тирозинемія типу І (тирозиноз). Причиною захворювання є, вірогідно, дефект ферменту фумарилацетоацетатгідролази, що каталізує розщеплення фумарилацетоацетату на фумарат і ацетоацетат (див. рис. 6.28). Накопичені метаболіти знижують активність деяких ферментів і транспортних систем амінокислот. Патофізіологія цього порушення досить складна. Гостра форма тирозинозу характерна для новонароджених. Клінічний прояв - діарея, блювання, затримки в розвитку. Без лікування діти гинуть у віці 6-8 місяців через недостатність печінки, що розвивається. Хронічна форма характеризується схожими, але менш вираженими симптомами. Загибель настає у віці 10 років. Уміст тирозину в крові хворих у декілька разів перевищує норму. Для лікування застосовують дієту зі зниженим вмістом тирозину і фенілаланіну.
Тирозинемія типу ІІ (синдром Ріхнера - Ханхорта). Причина - дефект ферменту тирозинамінотрансферази. Концентрація тирозину в крові хворих підвищена. Для захворювання характерні ураження очей і шкіри, помірна розумова відсталість, порушення координації рухів.
Тирозинемія новонароджених (короткочасна). Захворювання виникає в результаті зниження активності ферменту п-гідроксифенілпіруватдиоксигенази, що перетворює п-гідроксифенілпіруват у гомогентизинову кислоту (див. рис. 6.28). У результаті в крові хворих підвищується концентрація п-гідроксифенілацетату, тирозину й фенілаланіну. Для лікування призначають бідну на білок дієту і вітамін С.
Алкаптонурія (’’чорна сеча»). Причина захворювання - дефект диоксигенази гомогентизинової кислоти (рис. 6.28). Для цієї хвороби характерне виділення із сечею великої кількості гомогентизинової кислоти, яка, окиснюючись киснем повітря, утворює темні пігменти - алкаптони. Це метаболічне порушення було описане ще в XVI ст., а саме захворювання охарактеризоване в 1859 р. Клінічними проявами хвороби, крім потемніння сечі на повітрі, є пігментація сполучної тканини (охроноз) і артрит. Частота - 2-5 випадків на 1 млн новонароджених. Захворювання спадкується за аутосомно-рецесивним типом. Діагностичних методів виявлення гетерозиготних носіїв дефектного гену досі не розроблено.
Альбінізм. Причина метаболічного порушення - уроджений дефект тирозинази. Цей фермент каталізує перетворення тирозину на ДОФА в меланоцитах. У результаті дефекту тирозинази порушується синтез меланінів.
Клінічний прояв альбінізму (від лат. albus - білий) - відсутність пігментації шкіри й волосся. У хворих часто знижена гострота зору, виникає боязнь світла. Довге перебування таких хворих на відкритому сонці приводить до виникнення раку шкіри. Частота захворювання на альбінізм становить 1 : 20000.
Порушення синтезу катехоламінів (рис. 6.28) може викликати різні нервово-психічні захворювання, причому патологічні відхилення спостерігаються як при зниженні, так і при підвищенні їхньої кількості.
Хвороба Паркінсона. Захворювання розвивається внаслідок нестачі дофаміну в чорній субстанції мозку. Це одне з найпоширеніших неврологічних захворювань (частота - 1 : 200 серед людей віком понад 50 років). За цієї патології знижується активність тирозингідроксилази, ДОФА-декарбоксилази. Захворювання супроводжується трьома основними симптомами: акінезія (обмеженість рухів), ригідність (напруження м'язів), тремор (мимовільне дрижання). Дофамін не проникає через гематоенцефа- лічний бар'єр і як лікарський препарат не використовується. Для лікування паркінсонізму пропонують такі підходи:
✵ замісна терапія препаратами-попередниками дофаміну (похідними ДОФА) - леводопа, мадопар, наком та ін.;
✵ пригнічення інактивації дофаміну інгібіторами моноамінооксидази (депреніл, ніаламід, піразідол та ін.).
Депресивні стани часто пов'язані зі зниженням у нервових клітинах вмісту дофаміну й норадреналіну.
Гіперсекреція дофаміну в скроневій частці мозку спостерігається під час шизофренії.