Фізіологія людини - Вільям Ф. Ґанонґ 2002
Ендокринна система, метаболізм і репродуктивна функція
Енергетичний баланс, метаболізм і живлення
Метаболізм жирів
Ліпіди
Біологічно важливими ліпідами є жирні кислоти та їхні похідні, нейтральні жири (тригліцериди), фосфоліпіди і пов’язані з ними сполуки й стерини. Тригліцериди складаються з трьох жирних кислот, приєднаних до гліцерину (табл. 17-4). Жирні кислоти природного походження містять парну кількість атомів вуглецю. Вони можуть бути насиченими (не мають подвійних зв’язків) та ненасиченими (дегідрогенізовані, із різною кількістю подвійних зв’язків). Фосфоліпіди є складовими компонентами клітинних мембран. Стерини утворені з різних стероїдних гормонів та холестерину.
Окиснення жирних кислот та їхнє синтезування
В організмі жирні кислоти розпадаються до ацетил-КоА, який потрапляє до циклу лимонної кислоти. Головний розпад відбувається в мітохондріях шляхом ß-окиснення. Окиснення жирних кислот починається з їхнього активування (рис. 17-24) - реакції, що відбувається всередині й ззовні мітохондрій. Жирні кислоти із середнім та коротким ланцюгом надходять у мітохондрії без активування, тоді як жирні кислоти з довгим ланцюгом повинні бути приєднанні до карнітину естерним зв’язком, перш ніж вони зможуть пройти через внутрішню мітохондріальну мембрану. Карнітин - це ß-гідрокси-у-триметиламоній бутират, який синтезується в організмі з лізину і метіоніну. Транслоказа переміщає естер жирної кислоти і карнітину в матрикс мітохондрії в обмін на вільний карнітин. У матриксі естер переноситься на КоА, утворюючи активовану молекулу жирної кислоти, доступну для ß-окиснення, і постачаючи карнітин для подальшого обміну. Бета-окиснення полягає в повторному відщепленні фрагментів із двох атомів вуглецю від жирної кислоти (див. рис. 17-24). Енергетичний вихід цього процесу досить великий. Наприклад, катаболізм 1 моля шестивуглецевої жирної кислоти в циклі лимонної кислоти до СО2 та Н2О дає 44 молі АТФ, тоді як під час катаболізму шестивуглецевого вуглеводу глюкози утворюється лише 38 молів. Недостатнє ß-окиснення жирних кислот може бути спричинене нестачею карнітину чи генетичними дефектами в транслоказі або інших ензимах, задіяних у перенесенні жирних кислот із довгим ланцюгом у мітохондрії. Такий стан зумовлює кардіоміопатію, а також призводить до гіпокетонічної гіпоглікемії з комою - серйозним і часто смертельним станом, який виникає після голодування з вичерпанням запасів глюкози внаслідок недостатнього окиснення жирних кислот, яке постачає енергію, а кетонові тіла (див. нижче) не утворюються в достатніх кількостях з огляду на нестачу КоА в печінці.
Таблиця 17-4. Ліпіди
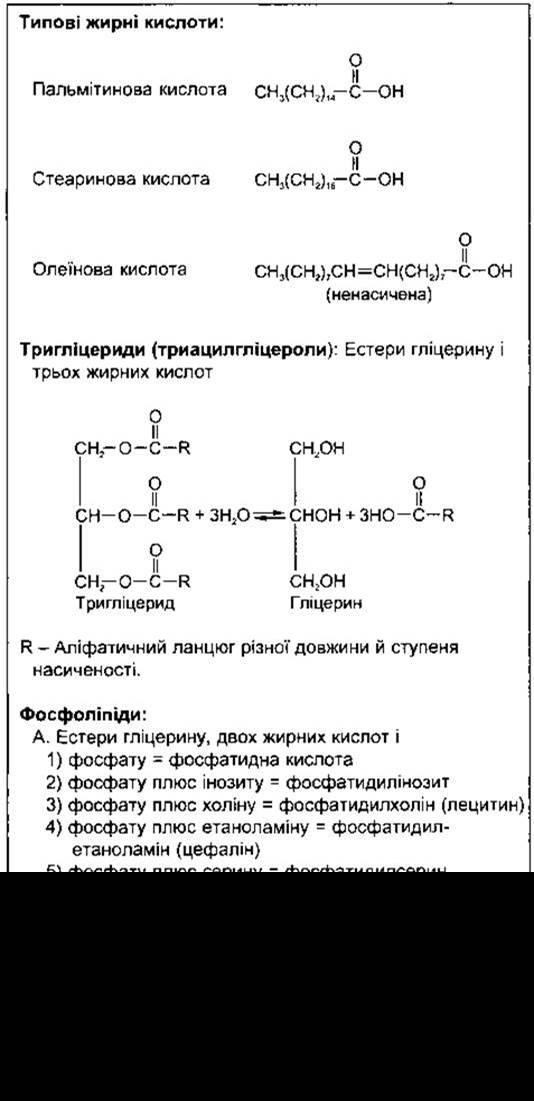
Багато тканин здатні синтезувати жирні кислоти з ацетил-КоА. Деякі синтези довголанцюгових жирних кислот із коротколанцюгових відбуваються в мітохондріях простим обігом реакцій, показаних на рис. 17-24. Однак здебільшого жирні кислоти синтезуються de novo з ацетил-КоА за допомогою різних шляхів, що розміщені головно поза мітохондріями в мікросомах. Етапи цих шляхів зображені на рис. 17-25.
З невідомих причин в усіх клітинах синтезування жирних кислот припиняється, коли довжина ланцюга сягає 16 вуглецевих атомів. Тільки невеликі кількості жирних кислот мають довжину 12- чи 14-вуглецевих атомів, і не утворюється жодної кількості кислот із довжиною вуглецевого ланцюга понад 16 атомів. Продовження вуглецевого ланцюга до 18 і більше атомів С (стеаринова кислота) відбувається у мікросомах печінкових клітин. Жирні кислоти сполучаються з гліцерином, утворюючи нейтральні жири, зокрема, цей процес відбувається у жирових відкладах, а саме з’єднання - в мітохондріях.
Кетонові тіла
У багатьох тканинах залишки ацетил-КоА конденсуються з утворенням ацетоацетил-КоА (рис. 17-26). У печінці, яка (на відміну від інших тканин) містить деацилазу, утворюється вільний ацетоацетат. Ця ß-кетокислота перетворюється в ß-гідроксибутират та ацетон, а оскільки ці сполуки погано метаболізують у печінці, то вони дифундують до кровоносної системи. Ацетоацетат у печінці виникає також унаслідок утворення 3-гідрокси-3-метилглутарил-КоА (див. рис. 17-26), і цей шлях є важливішим, ніж деацилювання. Ацетоацетат, ß-гідроксибутират та ацетон називають кетоновими тілами. Тканини, окрім печінки, переносять КоА із сукциніл-КоА до ацетоацетату та метаболізують «активний» ацетоацетат до СО2 і Н2О через цикл лимонної кислоти. Відомі й інші шляхи, за допомогою яких відбувається метаболізування кетонових тіл. За деяких умов кетони є важливим джерелом енергії. Ацетон виділяється із сечею та видихуваним повітрям.
Нормальний рівень кетонових тіл у крові людини низький (близько 1 мг/дл) і оскільки ці тіла за нормальних умов метаболізують настільки ж швидко, як і утворюються, то за 24 год їх екскретується менше 1 мг. Та якщо надходження ацетил-КоА до циклу лимонної кислоти зменшується внаслідок поганого постачання продуктами метаболізму глюкози чи не збільшується зі збільшенням постачання ацетил-, то ацетил-КоА нагромаджується, рівень конденсації ацетоацетил-КоА підвищується, і в печінці утворюється більше ацетоацетату. Здатність тканин до окиснення кетонових тіл швидко сягає межі, і ці тіла накопичуються в системі кровообігу (кетонемія). Два з трьох кетонових тіл - ацетоацетат і ß-гідроксибутират - є аніонами середньої сили кислот: ацетооцтової і ß-гідроксимасляної, вони мають буферні властивості, протидіючи відхиленню pH, що відбулося б в іншому випадку. Проте буферна ємність може бути перевищена, і метаболічний ацидоз, що розвивається за таких станів, як і діабетична кетонемія, буває важким і навіть фатальним.
До нестачі постачання внутрішньоклітинної глюкози призводять три стани: голодування, цукровий діабет та високожирова низьковуглеводна дієта. У разі діабету знижується надходження глюкози в клітину. Якщо ж більшість калорій їжі надходить із жирів, то виникає дефіцит вуглеводів, оскільки нема головного шляху перетворення жирів у вуглеводи. Клітини печінки наповнюються жиром, який їх ушкоджує й витісняє глікоген. За усіх цих станів кетонемія розвивається первинно з огляду на надмірне утворення кетонових тіл.
Запах ацетону у видихуваному повітрі у дітей після блювання виникає внаслідок кетонемії від голодування. Парентеральне введення порівняно малих кількостей глюкози запобігає утворенню кетонемії, саме тому цей вуглевод називають антикетогенним.
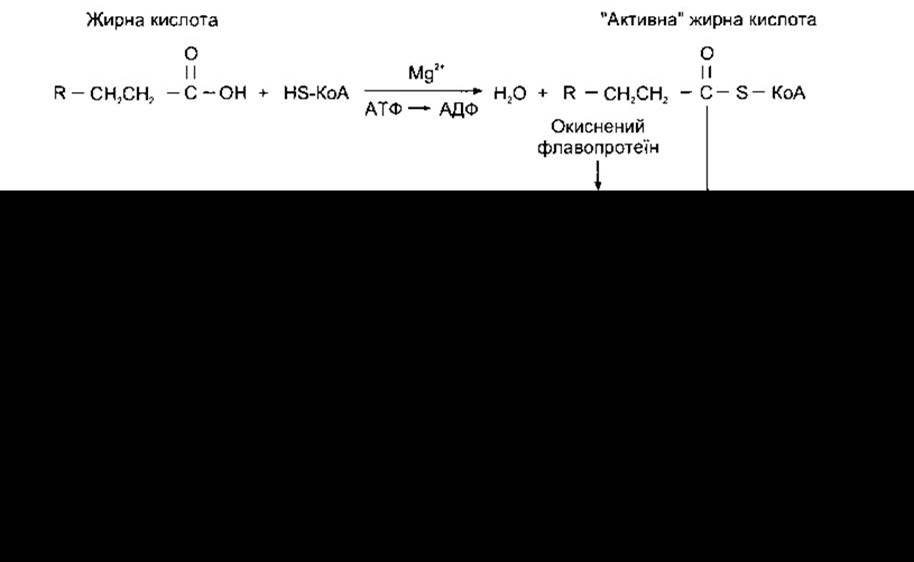
Рис. 17-24. Окиснення жирних кислот. Цей процес одночасного відщеплення фрагментів з двох атомів вуглецю повторюється до кінця ланцюга.
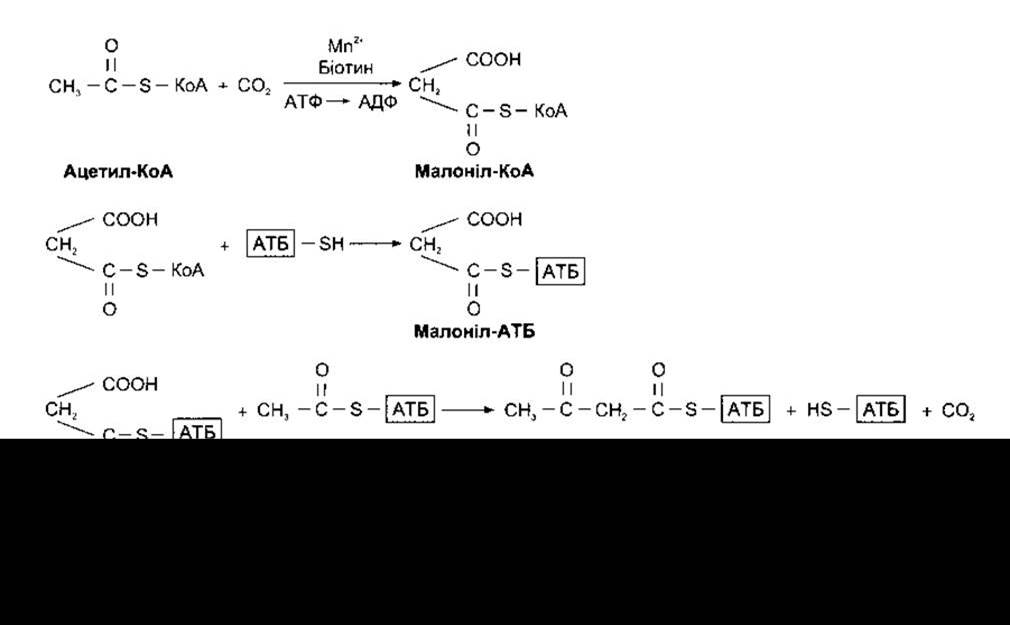
Рис. 17-25. Синтезування жирних кислот через шляхи, виявлені в мікросомах. АТБ - ацилтранспортний білок, який є частиною ензимного комплексу, до якого приєднуються ацильні залишки під час синтезування жирних кислот. Процес, показаний тут, повторюється, завдяки чому відбувається додавання одного комплексу ацетил-АТБ до бутирил-АТБ з утворенням шестивуглецевого АТБ похідного жирної кислоти, потім інший комплекс ацетил-АТБ з утворенням восьмивуглецевої сполуки і т.д.

Рис. 17-26. Утворення та метаболізм кетонових тіл. Зверніть увагу, що є два шляхи утворення ацетоацетату.
Клітинні ліпіди
Ліпіди в клітинах поділяють на два головні типи: структурні ліпіди, які є складовими мембран та інших частин клітини, та нейтральний жир, що його нагромаджують клітини-адипоцити у жирових відкладах. Нейтральний жир окиснюється під час голодування, а структурні ліпіди зберігаються. Звичайно ж, запаси жиру відрізняються за розмірами, проте в неопасистих осіб вони становлять 15% від маси тіла в чоловіків і 21 % - у жінок. Вони є не інертними масами, як уважали раніше, а активними динамічними тканинами, що зазнають постійного розпаду і ресинтезу. У жировій тканині глюкоза метаболізує до жирних кислот, і відбувається синтез нейтральних жирів. Відповідно, нейтральні жири розкладаються, і вільні жирні кислоти надходять у систему кровообігу.
Бурий жир
Третім - спеціальним - типом ліпідів є бурий жир, який становить лише малий відсоток від загальної кількості ліпідів організму. Бурий жир, який здебільшого простежується в дітей, однак буває і в дорослих, розташований між лопатками, на потилиці, вздовж великих судин у грудній клітці й черевній порожнині, а інколи і в інших місцях. У відкладах бурого жиру жирові клітини, як і кров’яні судини, мають виражену симпатичну іннервацію, на противагу відкладам білого жиру, у яких іннервуються лише деякі жирові клітини, проте основна симпатична іннервація є лише у кров’яних судинах. Окрім того, звичайні жирові клітини мають одну велику краплю білого жиру, тоді як клітини бурого жиру складаються з кількох маленьких крапель. Клітини бурого жиру містять також багато мітохондрій. У цих мітохондріях є звичайна внутрішня протонна провідність, яка утворює АТФ (окиснювальне фосфорилювання, див. вище), а також інша протонна провідність, що не утворює АТФ. Така закорочу вальна провідність залежить від роз’єднувального білка масою 32 кДа, тепер названого UCP 1 (від англ. uncoupling protein). Цей білок зумовлює роз’єднання окиснювального процесу та утворення АТФ, унаслідок чого утворюється більше тепла (рис. 17-27). Нещодавно відкрито другий білок - UCP 2, що міститься у білій та бурій жирових тканинах, є також докази наявності ще кількох таких білків. Активування симпатичної іннервації бурої жирової тканини вивільняє норадреналін, який, діючи на ß3-адpeнepгiчні рецептори, стимулює ліполіз, а посилене окиснення жирних кислот у мітохондріях збільшує теплопродукування. Отже, зміни активності симпатичної системи у бурому жирі спричинюють зміни в ефективності, із якою відбувається засвоювання їжі й утворюється енергія. Зміни в експресії білків UCP також можуть впливати на ефективність засвоєння їжі.
Є докази, що бурий жир діє таким способом у тварин і, можливо, людей, пристосованих до холоду, у яких посилено утворюється тепло в бурому жирі, а також пришвидшений кровообіг. Нервове стимулювання бурого жиру підвищене після споживання їжі, внаслідок чого теж збільшується утворення тепла. Зазначимо, що посилене утворення тепла після їжі зумовлене двома чинниками: негайно специфічною динамічною дією (див. вище) унаслідок засвоєння їжі, та дещо слабшим підвищенням вироблення тепла бурим жиром. Залежність кількості бурого жиру від споживання їжі розглянуто в Розділі 14.
Ліпіди плазми й транспортування ліпідів
Більшість ліпідів порівняно нерозчинні у воді і не циркулюють у вільній формі. Вільні жирні кислоти приєднані до альбуміну, тоді як транспортування холестерину, тригліцеридів та фосфоліпідів відбувається у вигляді ліпопротеїнових комплексів. Комплекси значно збільшують розчинність ліпідів. Є шість родин ліпопротеїнів (табл. 17-5) залежно від їхніх розмірів і вмісту ліпідів. Густина цих ліпопротеїнів (і, відповідно, швидкість седиментації в ультрацентрифузі) обернено пропорційна до вмісту в них ліпідів. Загалом ліпопротеїни мають гідрофобну серцевину з тригліцеридів і естерів холестеролу, оточених фосфоліпідами та білком (рис. 17-28). Шляхи, якими ліпопротеїни потрапляють із кишки до печінки (екзогенний шлях), і якими ліпіди переходять до та від тканин (ендогенний шлях), зображені на рис. 17-29. Білкові складові ліпопротеїнів називають апопротеїнами. Головні апопротеїни - АРО Е, АРО С та АРО В (див. рис. 17-29). Відомо дві форми АРО В: низькомолекулярна форма АРО В-48, притаманна екзогенному шляху, що транспортує екзогенні поглинуті ліпіди (див. нижче), та високомолекулярна форма АРО В-100, притаманна ендогенному транспортному шляху.
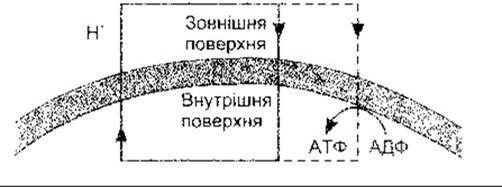
Рис. 17-27. Протонне транспортування через мітохондріальну мембрану у бурому жирі. Транспортування протонів назовні відбувається у електроннотранспортній системі, як і в інших мітохондріях; окрім руху протонів усередину, який сприяє утворенню АТФ, наявна «втеча» протонів усередину мітохондрії, яка не утворює АТФ. Унаслідок цього метаболізм жиру й утворення АТФ роз’єднані. Порівняйте з рис. 17-7
Хіломікрони утворюються в слизовій оболонці кишок під час абсорбції продуктів травлення жирів (див. Розділ 25). Вони є дуже великими ліпопротеїновими комплексами, що потрапляють у систему кровообігу через лімфатичні судини. Після споживання їжі в крові цих частинок так багато, що плазма набуває вигляду молока (ліпемія). Хіломікрони виводяться з кровообігу за участю ліпопротеїнліпази, яка розміщена на поверхні ендотелію. Цей ензим каталізує розпад тригліцеридів у хіломікронах до вільних жирних кислот та гліцерину, які потім потрапляють в адипоцити й реестерифікуються. Як альтернатива, вільні жирні кислоти залишаються в кров’яному руслі зв’язаними з альбуміном. Ліпопротеїнліпаза, яка потребує гепарин як кофактор, також видаляє тригліцериди з циркулювальних ліпопротеїнів дуже низької щільності (ЛПДНЩ) (див. нижче). Хіломікрони та ЛПДНЩ містять АРО С, - комплекс білків, що відділяються від них у капілярах. Один із компонентів комплексу - аполіпопротеїн С-ІІ - активує ліпопротеїнліпазу.
Таблиця 17-5. Головні ліпопротеїни. До ліпідів плазми належать ці компоненти плюс вільні жирні кислоти з жирових тканин, які циркулюють у зв’язаному з альбуміном стані
Ліпопротеїн |
Розмір, нм |
Склад, % |
Походження |
||||
білок |
вільний холестерол |
естери холестеролу |
тригліцерид |
фосфоліпіди |
|||
Хіломікрони |
75-100 |
2 |
2 |
3 |
90 |
3 |
Кишка |
Залишки хіломікронів |
30-80 |
... |
... |
... |
... |
... |
Капіляри |
Ліпопротеїни дуже низької щільності |
30-80 |
8 |
4 |
16 |
55 |
17 |
Печінка та кишка |
Ліпопротеїни середньої щільності |
25-40 |
10 |
5 |
25 |
40 |
20 |
ЛПДНЩ |
Ліпопротеїни низької щільності |
20 |
20 |
7 |
46 |
6 |
21 |
ЛПСЩ |
Ліпопротеїни високої щільності |
7,5-10 |
50 |
4 |
16 |
5 |
25 |
Печінка та кишка |
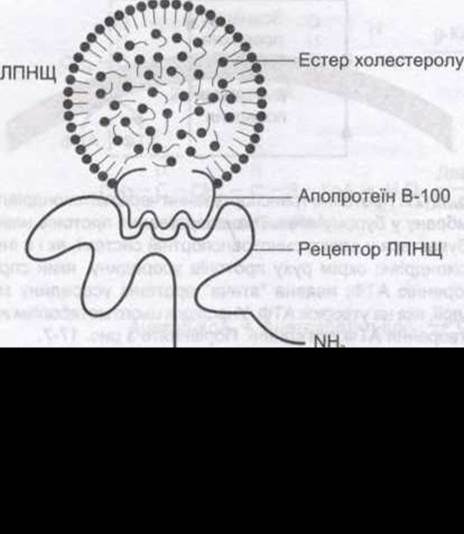
Рис. 17-28. Схематичне зображення структури ліпопротеїну низької щільності, рецептора ЛПНЩ та зв'язування ЛПНЩ з рецептором через АРО В-100.
Позбавлені тригліцеридів хіломікрони залишаються в кров’яному руслі як багаті на холестерол ліпопротеїни, названі хіломікронними залишками, що мають 30-80 нм у діаметрі. Ці залишки потрапляють до печінки, де приєднуються до хіломікронних залишків та рецепторів ЛНЩ, і починається внаслідок рецепторопосередкованого ендоцитозу (див. Розділ 1) розщеплення в лізосомах.
Хіломікрони та їхні залишки є транспортною системою для екзогенних ліпідів їжі (див. рис. 17-29). Наявна також ендогенна система, що складається з ЛПДНЩ, ліпопротеїнів середньої щільності (ЛПСЩ), ліпопротеїнів низької щільності (ЛПНЩ) та ліпопротеїнів високої щільності (ЛПВЩ), які транспортують тригліцериди і холестерол в організмі. ЛПДНЩ утворюються в печінці і транспортують тригліцериди, сформовані з жирних кислот та вуглеводів, до периферійних тканин. Якщо багато тригліцеридів, то вони перетворюються в ЛПСЩ, які віддають фосфоліпіди і під дією плазматичного ензиму лецитин-холестеролацилтрансферази (ЛХАТ; див. рис. 17-29) приєднують естери холестерольного і ЛПВЩ походження. Деякі ЛПСЩ видаляє печінка; інші втрачають тригліцериди та білок, очевидно, в синусоїдах печінки, і перетворюються в ЛПНЩ. Під час цього перетворення вони втрачають АРО Е, проте АРО В-100 залишається.
ЛПНЩ постачають тканинам холестерол - важливу складову клітинних мембран, яка в специфічних залозах використовується для утворення стероїдних гормонів. У печінці та більшості позапечінкових тканин відбувається поглинання ЛПНЩ рецепторопосередкованим ендоцитозом в облямованих ямках (див. Розділ 1). Рецептори розпізнають компонент ЛПНЩ - АРО В-100 (див. рис. 17- 28); вони також зв’язуються з АРО Е, але не з АРО В-48.
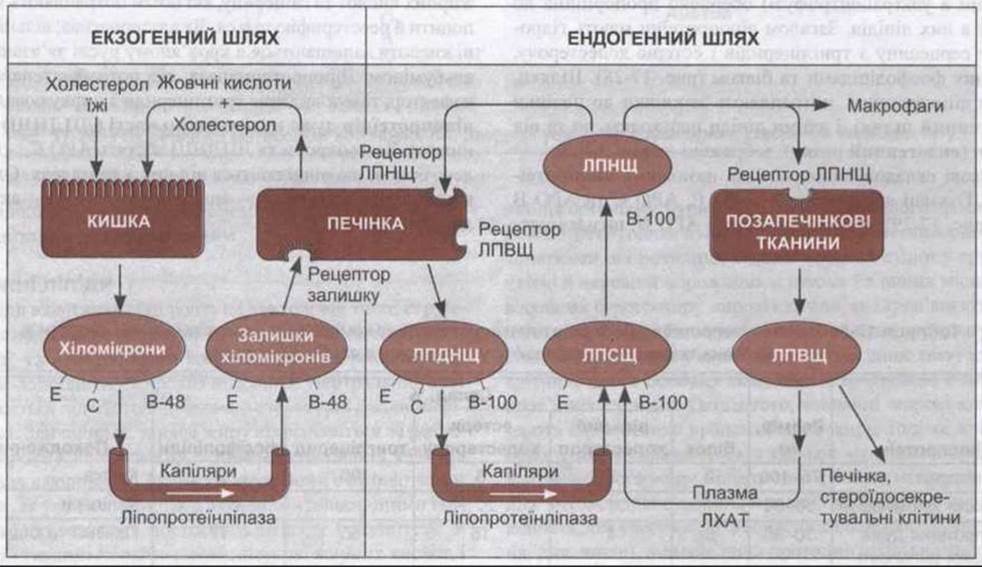
Рис. 17-29. Спрощена схема ліпопротеїнової системи транспортування ліпідів у людини. В екзогенній системі хіломікрони, багаті на тригліцериди, що надходять з їжі, під дією ліпопротеїнліпази перетворюються в залишки хіломікронів, багаті на естери холестеролу. В ендогенній системі печінка секретує ЛПДНЩ, багаті на тригліцериди, вони перетворюються в ЛПСЩ, а потім - у ЛПНЩ, багаті на естери холестеролу. ЛХАТ - лецитинхолестеролацилтрансфераза. Букви на хіломікронах, залишках хіломікронів, ЛПДНЩ, ЛПСЩ, ЛПНЩ позначають головні апопротеїни, знайдені в них. Одну третину ЛПСЩ поглинають макрофаги та інші клітини за допомогою альтернативних механізмів.
Рецептори ЛПНЩ людини належать до родини рецепторів, які транспортують макромолекули всередину клітини шляхом ендоцитозу у клатриноблямованих ямках (див. Розділ 1). Це велика комплексна молекула, в якій є багата на цистеїн ділянка з 292 амінокислотних залишків, що приєднується до ЛПНЩ; ділянка з близько 400 амінокислотних залишків, що є гомологічною до попередника епідермального фактора росту; ділянка з 58 амінокислот, що багата на серин і треонін та є місцем глікозилювання; ділянка з 22 гідрофобних амінокислотних залишків, що пронизує клітинну мембрану; ділянка з 50 амінокислотних залишків, що виходять у цитоплазму (див. рис. 17-28). Ген цього білка містить 18 екзонів, 13 із яких кодують білкові послідовності, що є гомологічними до послідовностей в інших білках. Отже, рецептор ЛПНЩ є мозаїчним білком, утвореним з екзонів, які кодують також інші білки.
У процесі рецепторопосередкованого ендоцитозу кожна облямована ямка відділяється з утворенням облямованого пухирця, а потім - ендосоми. Ионоселективні помпи в мембранах ендосом знижують pH у цих органелах. У випадку рецептора ЛПНЩ, але не рецептора залишків хіломікронів, це ініціює вивільнення рецепторів ЛПНЩ, які інкорпоруються у клітинну мембрану (рис. 17-30). Після цього ендосоми зливаються з лізосомами, і холестерол, утворений з естерів холестеролу під дією кислої ліпази в лізосомах, стає придатним для використання на клітинні потреби (див. рис. 17-30). Холестерол у клітинах гальмує внутрішньоклітинне синтезування холестеролу шляхом інгібування ГМГ-КоА редуктази (див. нижче) та стимулює естерифікацію надлишку вивільненого холестеролу й інгібує синтезування нових рецепторів ЛПНЩ. Усі ці реакції забезпечують механізм зворотного контролювання за кількістю холестеролу в клітинах.
Виділення ЛПНЩ також поглинає система з нижчою спорідненістю в макрофагах і деяких інших клітинах. Окрім того, макрофаги переважно поглинають ЛПНЩ, змінені внаслідок окиснення. Проте окиснення може також відбуватись у макрофагах. У тварин великі дози антиоксидантів, таких як вітамін Е, сповільнюють прогресування атеросклерозу, однак досі результати подібних дослідів у людей незадовільні. Рецептори ЛПНЩ на макрофагах та споріднених клітинах називають очисними рецепторами. Вони відмінні від рецепторів на інших клітинах і більше споріднені зі зміненими ЛПНЩ. Якщо макрофаги перевантажені окисненими ЛПНЩ, то вони перетворюються в «пінисті клітини», які можна виявити в уражених раннім атеросклерозом судинах. За стабільних умов холестерол проникає в клітини та виходить із них. Уважають, що він виводиться з клітин одним із білків ABC-касети (див Розділ 1), і його поглинають ЛПВЩ. Ці ліпогіротеїни синтезуються в печінці та кишці. Індивідуальний рецептор ЛПВЩ виявлений і клонований. Спершу його виявили в ендокринних залозах, що утворюють стероїдні гормони, та в печінці. Система ЛПВЩ переносить холестерол у печінку, після чого він екскретується в жовч. У цьому випадку відбувається зниження вмісту холестеролу в плазмі.
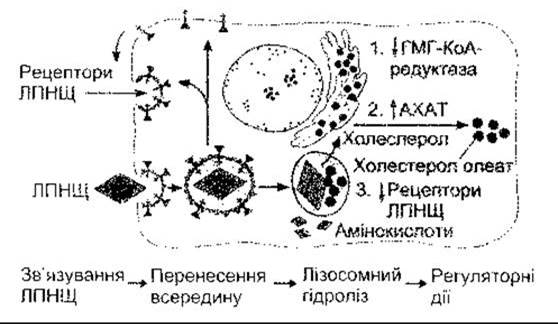
Рис. 17-30. Поглинання клітиною холестеролу та його метаболізм. ЛПНЩ зв’язуються з рецепторами та переходять завдяки рецепторопосередкованому ендоцитозу в ендосоми з низьким pH. Рецептори вивільняються і рециркулюють у мембрану. Естери холестеролу потрапляють у лізосоми, де вивільняється холестерол, потрібний для клітинних процесів. Холестерол також інгібує ГМГ-КоА-редуктазу (1), потім його частково перетворює в інший естер холестеролу ензим ацетил-КоА (2): холестеролацилтрансфераза (АХАТ), та інгібує утворення рецепторів ЛПНЩ (3) (з дозволу MS Brown).
Апопротеїн Е синтезують клітини мозку, селезінки, легень, надниркових залоз, яєчників та нирок, а також печінки. Його концентрація значно збільшується в разі ушкодження нервів, адже він бере участь у їхньому відновленні. Ген аполіпопротеїну Е наявний у популяції у трьох алелях: АРО-2, АРО-3 та АРО-4. Алель АРО-4 менше поширений, ніж АРО-2 і АРО-3, однак більше поширений у пацієнтів із хворобою Альцгеймера (див. Розділ 16), і, мабуть, причетний до її розвитку.
Метаболізм вільних жирних кислот
Вільні жирні кислоти (ВЖК) надходять у жирові клітини та інші тканини завдяки хіломікронам та ЛПДНЩ (див. вище). їхнє синтезування відбувається також у жирових відкладах, де вони зберігаються. ВЖК циркулюють, будучи зв’язаними з альбуміном, і є головним джерелом енергії багатьох органів. їх інтенсивно засвоює серце, крім того, можливо, усі тканини, у тім числі мозок, можуть окиснювати ВЖК до СО2 та Н2О.
Постачання тканинам ВЖК регульоване двома ліпазами. Як зазначено, ліпопротеїнліпаза на поверхні ендотелію капілярів гідролізує тригліцериди в хіломікронах та ЛПДНЩ, унаслідок чого утворюються ВЖК та гліцерин, які знову з’єднуються в нові тригліцериди в жирових клітинах. Внутрішньоклітинна гормоночутлива ліпаза адипоцитів каталізує розпад акумульованих тригліцеридів на гліцерин та жирні кислоти, після чого вони потрапляють у систему кровообігу.
Гормоночутлива ліпаза перетворюється з неактивної в активну форму за допомогою цАМФ через протеїнкіназу А (рис. 17-31). Аденілатциклазу в адипоцитах, відповідно, активує глюкагон, а також катехоламіни (норадреналін та адреналін) через ß-адренергічний рецептор. Реакції цього рецептора атипові; наприклад, він резистентний до анаприліну та інших препаратів, що блокують ß1- та ß2-peцeптори, і є, можливо, ß3-адрeнергічним рецептором. Тривають спроби розробити селективний ß3-arOHiCT для можливого використання в лікуванні ожиріння. АКТГ, ТТГ, ЛГ, серотонін та вазопресин збільшують ліполіз через цАМФ, однак фізіологічна роль цих речовин у регулюванні ліполізу остаточно не з’ясована.
Гормон росту, глюкокортикоїди та тиреоїдні гормони також збільшують активність гормоночутливої ліпази, проте завдяки сповільненню процесу синтезування нового білка. Припускають, що гормон росту стимулює утворення білка, що збільшує здатність катехоламінів активувати цАМФ, тоді як кортизол сприяє утворенню білка, що підсилює дію цАМФ. З іншого боку, інсулін та простагландин Е зменшують активність гормоночутливої ліпази, можливо, гальмуючи утворення цАМФ.
Активність гормоночутливої ліпази збільшується під час голодування та стресу і зменшується у разі споживання їжі та інсуліну. Харчування збільшує, а голодування та стрес, навпаки, зменшують активність ліпопротеїнліпази.
Метаболізм холестеролу
Попередником стероїдних гормонів та жовчних кислот є холестерол - як уже зазначено, важливий складовий компонент клітинних мембран (див. Розділ 1). Його виявлено тільки у тварин. Подібні стерини є у рослин, проте вони за нормальних умов не поглинаються в шлунково-кишковому тракті. Більшість холестеролу їжі міститься в яєчних жовтках і тваринному жирі. Поглинання холестеролу відбувається в кишці, де він інкорпорується в хіломікрони, що утворюються в слизовій оболонці. Після того, як хіломікрони в жировій тканині звільняються від тригліцеридів, залишки хіломікронів постачають холестерол у печінку. Печінка та інші тканини також синтезують холестерол. Частина холестеролу в печінці екскретується в жовч як у вільній формі, так і у вигляді жовчних кислот, і реабсорбується в кишках. Більшість холестеролу в печінці входить у ЛПДНЩ і циркулює у ліпопротеїнових комплексах (див. вище).
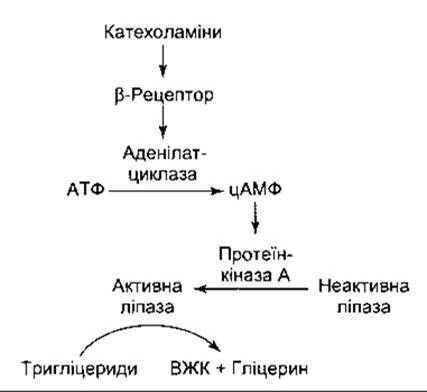
Рис. 17-31. Механізм, за допомогою якого катехоламіни збільшують активність гормоночутливої ліпази в жировій тканині. Бета-рецептор є, можливо, Р3-адгенергічним рецептором.
Біосинтез холестеролу з ацетату показано на рис. 17- 32. Холестерол за допомогою механізму зворотного зв’язку гальмує своє синтезування шляхом інгібування ГМГ-КоА- редуктази - ензиму, що перетворює 3-гідроксиметилглутарил-коензим А у мевалонову кислоту. Отже, якщо споживання холестеролу з їжею високе, то зменшується синтезування холестеролу в печінці, і навпаки. Проте компенсація за допомогою механізму зворотного зв’язку неповна, оскільки їжа з малим умістом холестеролу і насичених жирів призводить лише до незначного зниження рівня холестеролу в крові.
Рівень холестеролу в плазмі знижується під дією тиреоїдних гормонів, які збільшують кількість рецепторів ЛПНЩ у печінці, та під дією естрогенів, які підвищують концентрацію ЛПВЩ у плазмі та знижують рівень ЛПНЩ. Естрогени посилюють катаболізм ЛПНЩ, що циркулюють, ймовірно, шляхом збільшення кількості рецепторів ЛПНЩ у печінці. Рівень холестеролу в плазмі підвищується в разі закупорення жовчних шляхів та нелікованого цукрового діабету. Якщо реабсорбцію жовчних кислот у кишці послаблюють смоли, такі як колестипол, то для утворення жовчних кислот потрібно більше холестеролу. Однак зниження рівня холестеролу в плазмі є порівняно малим унаслідок компенсаторного збільшення синтезу холестеролу. Ловастатин та близькі статини гальмують синтезування холестеролу шляхом прямого інгібування ГМГ-КоА-редуктази (див. рис. 17-32). Іншим препаратом, який широко використовують для зниження рівня холестеролу в плазмі, є вітамін ніацин; він у великих дозах інгібує мобілізацію вільних жирних кислот з периферійних відкладів жиру, а отже, зменшує синтезування ЛПДНЩ у печінці. Ще один препарат - клофібрат - діє комплексно збільшуючи окиснення жирних кислот у печінці та м’язах і зменшуючи секрецію ліпопротеїнів у печінці. Незважаючи на це, сьогодні статини є загальновідомими широко використовуваними високоефективними препаратами.
Зв’язок з атеросклерозом
Холестерол має важливе значення в етіології та патогенезі атеросклерозу. Ця винятково поширена хвороба спричинює розвиток інфаркту міокарда, церебрального тромбозу, ішемічної гангрени кінцівок та інших серйозних захворювань. Вона виникає внаслідок інфільтрації холестеролу та появи пінистих клітин в уражених артеріальних стінках. Це супроводжується складним каскадом змін, що охоплює тромбоцити, макрофаги, гладкі м’язові клітини та фактори росту, що утворюють проліферувальні ушкодження, які врешті-решт кальцифікуються. Ці зміни призводять до деформування судин і роблять їх ригідними. Нефахівці зачислюють цей стан до артеріосклерозу, однак формально артеріосклероз є загальнішим поняттям, що означає втрату еластичності чи затвердіння артерій з будь-якої причини. В осіб із підвищеним рівнем холестеролу це є причиною розвитку атеросклерозу та його ускладнень. Нормальний рівень холестеролу в плазмі становить 120- 200 мг/дл, проте в чоловіків простежується чіткий тісний позитивний взаємозв’язок між рівнем смертності від ішемічної хвороби серця й рівнем холестеролу в плазмі (понад 180 мг/100 мл). Крім того, тепер відомо, що зниження рівня холестеролу в плазмі шляхом дієти чи вживання ліків сповільнює і навіть спричинює регресію атеросклеротичних ушкоджень та ускладнень, зумовлених ними. Цікаво, що зменшення кількості інфарктів міокарда безпосередньо корелює зі ступенем звуження коронарних артерій. Доведено також, що зниження рівня холестеролу в плазмі може запобігти розриву атеросклеротичних бляшок, навіть таких, що часто ініціюють утворення тромбів (див. Розділ. 32).
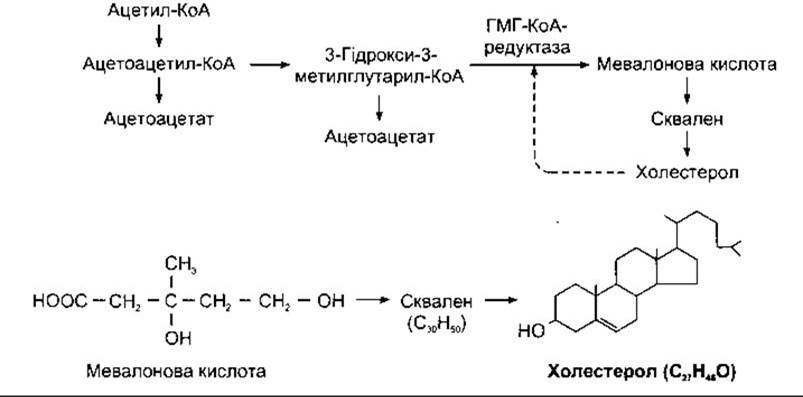
Рис. 17-32. Біосинтез холестеролу. Шість молекул мевалонової кислоти конденсують з утворенням сквалену, який потім гідроксилює та перетворюється в холестерол. Штрихова стрілка показує інгібування холестеролом завдяки регулюванню механізму зворотного зв’язку, ГМГ-КоА-редуктази, ензиму, що каталізує утворення мевалонової кислоти.
Рівень холестеролу в плазмі підвищується під час споживання їжі та за деяких станів, у тім числі родинної гіперхолестеролонемії внаслідок різноманітних мутацій рецептора ЛПНЩ. Під час обстеження пацієнтів, окрім рівня холестеролу, варто також аналізувати рівні ЛПНЩ та ЛПВЩ у плазмі. Особи із збільшеним рівнем ЛПНЩ мають вищий, ніж звичайно, рівень захворюваності та ускладнень, тоді як особи з підвищеним рівнем ЛПВЩ - навпаки. Цікаво, що жінки, серед яких рівень захворюваності на інфаркт міокарда є нижчим, ніж серед чоловіків, мають вищі рівні ЛПВЩ. Окрім того, рівень ЛПВЩ підвищується в тренованих осіб і тих, що випивають одну чи дві порції алкогольних напоїв (100 мл) денно, а знижується в осіб, що курять, є огрядними чи ведуть сидячий спосіб життя. Помірне споживання алкоголю зменшує частоту інфарктів міокарда, а повнота та куріння - це фактори ризику, що її збільшують.
Є докази, що підвищені рівні ЛПСЩ, залишків хіломікронів та тригліцеридів також призводять до атеросклерозу, тоді як підвищені рівні ЛПДНЩ та хіломікронів - ні. Інші фактори, що сприяють атеросклерозу, розглянуто в Розділі 32.
Незамінні жирні кислоти
Тварини, що не отримують жирів із їжею, перестають рости, в них розвиваються ушкодження шкіри та нирок, вони стають безплідними. Додавання ліноленової, лінолевої та арахідонової кислот до їхнього раціону дає змогу вилікувати всі симптоми дефіциту. Ці три кислоти є поліненасиченими жирними кислотами і завдяки їхній дії їх називають незамінними жирними кислотами. Подібні дефіцитні симптоми не зафіксовано однозначно у людей, однак є причини вважати, що деякі ненасичені жири - незамінні складові їжі, зокрема, в дітей. В організмі відбувається дегідрогенація жирів, проте не синтезуються вуглецеві ланцюги з таким розташуванням подвійних зв’язків, як у незамінних жирних кислот.
Ейкозаноїди
Одна з причин, а можливо і єдина, того, що незамінні жирні кислоти необхідні для здоров’я, - вони є попередниками простагландинів, простациклінів, тромбоксанів, ліпоксинів, лейкотрієнів та близьких сполук (рис. 17-33). Ці сполуки називають ейкозаноїдами, бо вони походять від 20-вуглецевої (ейкоза-) поліненасиченої жирної кислоти - арахідонової кислоти (арахідонату), і 20-вуглецевих похідних лінолевої та ліноленової кислот. Зазначимо, що вони утворюються з арахідонової кислоти за допомогою трьох окремих груп ензимів (див. рис. 17-33).
Простагландини - це ряд 20-вуглецевих ненасичених жирних кислот, що містять циклопентанове кільце. Спершу їх виділили з передміхурової залози, однак тепер з’ясовано, що вони синтезуються в більшості, а можливо, й усіх органах тіла. Структура деяких з них показана на рис. 17- 34.
Простагландини (ПГ) поділяють на групи - наприклад, на ПГЕ та ПГF - за конфігурацією циклопентанового кільця. Кількість ненасичених зв’язків у бічних ланцюгах позначає нижній індекс, наприклад, простагландин групи Е, зображений на рис. 17-34, є простагландином ПГЕ2.

Рис. 17-33. Головні біологічно активні сполуки, що утворюються з арахідонової кислоти; ГЕТЄ - гідроксиейкозатетраєнова кислота; ДГТ - дигідроксиейкозатетраєнова кислота; ЕЕТ - епоксиейкозатриєнова кислота.
Попередником різноманітних інших простагландинів, тромбоксанів та простацикліну є простагландин Н2 (ПГН2). У цьому синтезі (див. рис. 17-34) задіяні різні ензими. Утворюється ПГН2 з арахідонової кислоти циклооксигеназами (СОХ - від англ. cyclooxygenase). Є дві ізоформи циклооксигеназ, що кодовані різними генами: циклооксигеназа-1 (СОХ-1) та циклооксигеназа-2 (СОХ-2): СОХ- 1 експресована постійно, тоді як СОХ-2 індукована факторами росту, цитокінами та активаторами росту пухлин.
Відомими рецепторами простагландинів є серпентинові рецептори, що діють через гетеротримерні G-білки. Виділено чотири рецептори ПГЕ2, ЩО отримали відповідні назви - від ЕР 1 до ЕР 4 (табл. 17-6), а також простациклінові та рецептор ПГЕ2а.
Ефекти дії простагландинів різноманітні. Багато з них описано в розділах, що присвячені тим системам, у яких вони відіграють важливу роль. Вони особливо важливі в жіночому репродуктивному циклі, під час пологів, у серцево-судинній системі, у разі запальної відповіді та виникненні болю. Нові докази свідчать, що вони беруть участь у канцерогенезі, регулюванні апоптозу та ангіогенезу. Тромбоцити синтезують тромбоксан А2, який спричинює агрегацію тромбоцитів та є судинозвужувальним агентом. Рецептор тромбоксану А - це типовий серпентиновий рецептор, з’єднаний з G-білком (див. табл. 17-6); він діє через фосфатидилінозитол, який відкриває Са2+-активовані канали Сl. Тромбоксан В2 є метаболітом тромбоксану А2. Простациклін же інгібує агрегацію тромбоцитів і є судинорозширювальним агентом. Його утворюють клітини ендотелію та гладких м’язів у стінках кровоносних судин. Вивільнення тромбоксану А2 у кров, яке забезпечують тромбоцити, у місці безпосереднього ушкодження спричинює утворення тромбу, тоді як вивільнення простацикліну в сусідніх ділянках судин приводить до локалізації тромбу і підтримує прохідність інших ділянок судин. Оскільки дія аспірину на тромбоцити триваліша, ніж дія на стінки кровоносних судин, то він зумовлює антикоагуляційний ефект, цінний для запобігання міокардіальним ускладненням та інсульту (див. Розділ 31).
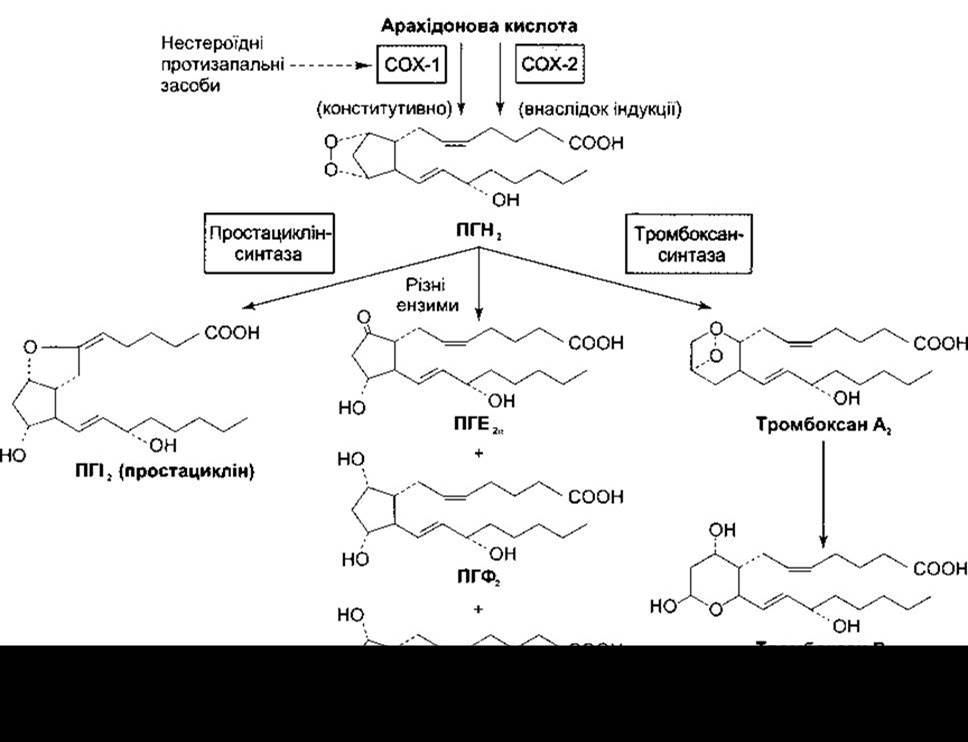
Рис. 17-34. Метаболізм арахідонової кислоти за участю цикпооксигенази-1 (СОХ-1) та цикпооксигенази-2 (СОХ-2).
Арахідонова кислота також перетворюється в 5-гідропероксиейкозатетраєнову кислоту (5-ГПЕТЄ) та 5-ліпоксигеназу, що беруть участь в активуванні 5-ліпоксигеназоактивувального білка (ПЛАБ). 5-ГПЕТЄ перетворюється в лейкотрієни (LT - від англ. leukotrienes). Чотири лейкотрієни є аміноліпідами, що містять амінокислоти; лейкотрієн С4 (LTC4) МІСТИТЬ три пептид глутатіон, LTD4 - гліцин та цистеїн, LTE4 - цистеїн, a LTF4 - цистеїн та глутамінову кислоту (рис. 17-35). Окрім того, арахідонова кислота перетворюється в ліпоксини через 15-ГПЕТЄ (див. рис. 17-35).
Лейкотрієни, тромбоксани, ліпоксини та простагландини називають місцевими гормонами. Вони мають короткий період півжиття, інактивуються в багатьох різних тканинах і, без сумніву, діють у тих тканинах, де й утворюються.
Протизапальні стероїди, такі як кортизол, за допомогою фосфоліпази А2 гальмують вивільнення арахідонової кислоти з її фосфоліпідних запасів, а отже, сповільнюють утворення усіх її похідних (див. рис. 17-33). Нестероїдні протизапальні засоби (НСПЗЗ), такі як аспірин та індометацин, інгібують циклооксигенази, не торкаючись шляхів каталізу ліпоксигенази та CYP-ензимів (див. рис. 17-34). Інгібування СОХ-1 в експериментальних тварин спричинює шлунково-кишкові та ниркові аномалії, тоді як інгібування СОХ-2 послаблює біль, запалення та лихоманку. Аспірин, індометацин та більшість інших клінічно доступних НСПЗЗ блокують одночасно і СОХ-1, і СОХ-2, однак важливою є розробка COX-2-інгібіторів із метою зменшення шлунково-кишкових та ниркових побічних ефектів у разі застосування названих препаратів для лікування болю, запалень та артриту.
Лейкотрієни є трансмітерами алергічної відповіді та запалення. їхнє вивільнення спричинене приєднанням специфічних алергенів до IgE-антитіл на поверхні гладких м’язових клітин. Лейкотрієни зумовлюють бронхоконстрикцію, звужують артеріоли, збільшують судинну проникність та притягують нейтрофіли й еозинофіли до місць запалень. У нокаутних мишей зі зруйнованим геном 5-ліпоксигенази розвиток та загальний стан нормальні, однак є резистентність до деяких форм запалення. У людей розвиток астми, псоріазу синдрому розладу дихання в дорослих, алергічного риніту, хвороби Крона та виразкового коліту пов’язують з нокаутом такого гена.
Таблиця 17-6. Рецептори простагландинів та тромбоксанів
ПГІ2 |
IP |
ПГЕ2 |
ЕР 1, ЕР2, ЕРЗ, ЕР 4 |
ПГF2a |
FP |
Тромбоксан А |
ТР |
Два рецептори лейкотрієнів, що містять цистеїн, - CysLT1 та CysLT2, схарактеризовані фармакологічно, хоча їхні структури і досі невідомі. Рецептор лейкотрієну В4 - ВЛТ - є серпентиновим рецептором, з’єднаним із G-білком. Рецептор CysLT1, опосередковує бронхоконстрикцію, хемотаксис та підвищену судинну проникність, рецептор CysLT2 - скорочення гладких м’язів судин легенів, а рецептор BLT - переважно хемотаксис. Зв’язок цих рецепторів з астмою розглянуто в Розділі 37.
Ліпоксин А розширює малі судини, ліпоксин А та ліпоксин В зменшують цитотоксичний ефект справжніх клітин-убивць (див. Розділ 27). Проте їхнє фізіологічне значення досі не з’ясоване.
Зазначимо, що 12-ГЕТЄ, кілька дигідроксипохідних ейкозатетриєнової кислоти (ДГТ) та кілька епоксиейкозатриєнових кислот (ЕЕТ) утворені з арахідонової кислоти цитохром Р450 (CYP) монооксигеназами (див. рис. 17- 33). Роль цих продуктів невідома, однак ДГТ та ЕЕТ впливають на екскрецію нирками солей та води, що може мати важливе фізіологічне значення. Цитохром Р450 є дивовижною групою з понад 300 ензимів, що каталізують окиснення, епоксидування, аліфатичне гідроксилювання та інші реакції. У ссавців вони задіяні не лише в метаболізмі ейкозаноїдів, а й у синтезі стероїдних гормонів, метаболізмі ліків та окисненні жирних кислот. Їх розподілено на родини та підродини за гомологічною послідовністю (CYP1, CYP2, CYP3 і т.д.), у людини виявлено 12 родин.
Ожиріння
Ожиріння - найпоширеніший та найдорожчий з харчових розладів у країнах з високим розвитком. Воно вже вразило 33% дорослого населення і поширюється далі. Пов’язують його з підвищеним ризиком атеросклерозу, діабету та хвороб жовчного міхура. Нормальні відклади жиру становлять 12-18% від маси тіла в чоловіків і 18-25% - у жінок. Про ожиріння говорять, коли ці значення перевищують 20% у чоловіків та 25% у жінок. Також широко використовують таблицю норм росту та маси. Однак показником, що найліпше пов’язаний з жиром тіла, є індекс маси тіла; він дорівнює масі тіла (у кілограмах), поділеній на квадрат зросту (у метрах). Нормальне значення цього індексу становить 20-25 кг/м2. Маса тіла зростає і є стабільною під час третього десятиліття життя, потім дещо збільшується, а у похилому віці зменшується. Якщо споживання їжі з віком не зменшується, то виникає ожиріння. Окрім того, із віком знижується основний обмін речовин.
У людей простежується виражений генетичний компонент в ожирінні, однак на нього також впливають і чинники навколишнього середовища. Наприклад, у США ожиріння серед жінок з вищих соціально-економічних груп трапляється набагато рідше, ніж серед жінок із нижчих груп. Зв’язок ожиріння з діабетом розглянуто в Розділі 19, а з контролем споживання їжі - у Розділі 14.
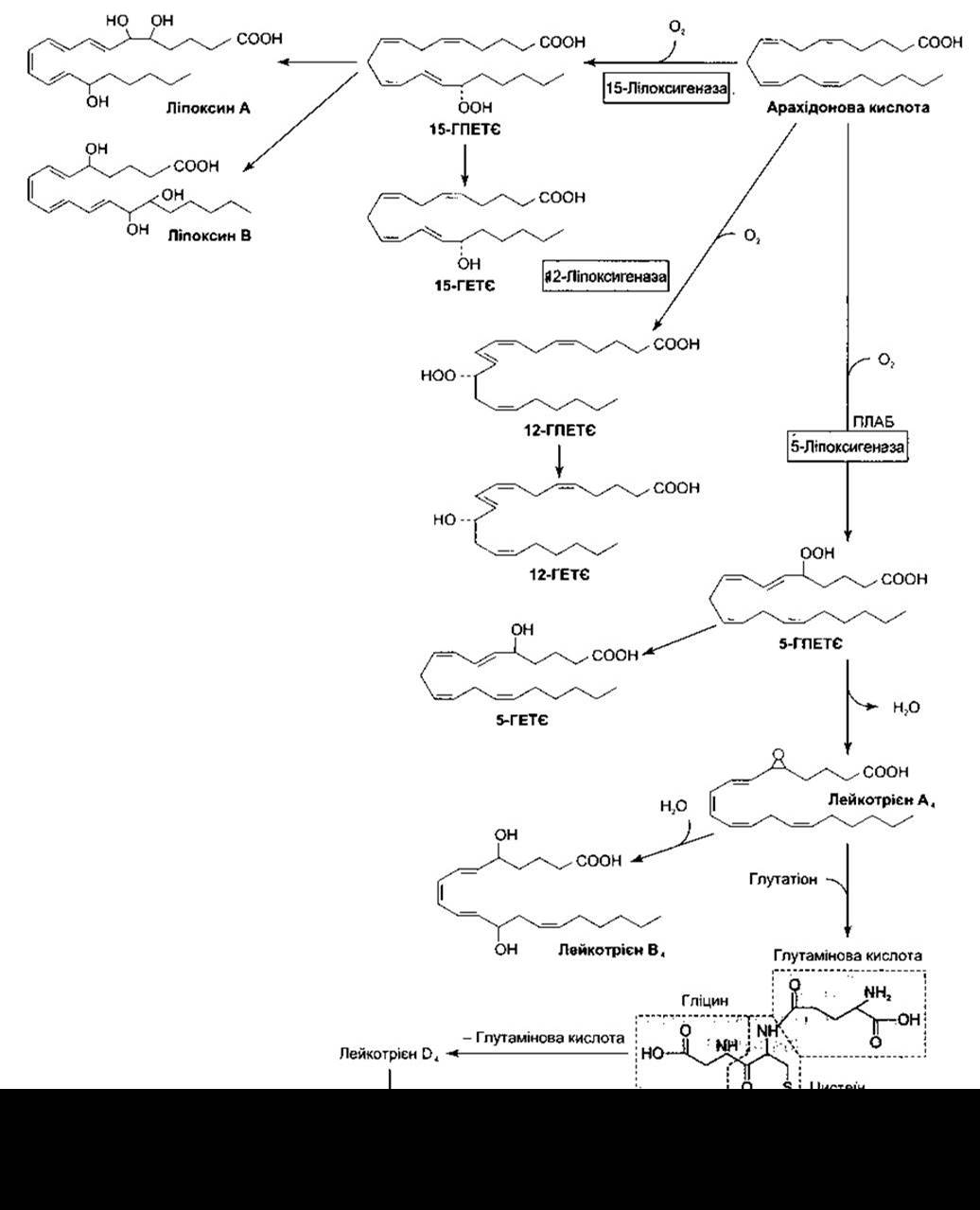
Рис. 17-35. Метаболізм арахідонової кислоти ліпоксигеназами; ПЛАБ - 5-ліпоксигеназоактивувальний білок.