Молекулярная биология клетки - Том 1 - Албертс Б., Брей Д., Льюис Дж., Рэфф М., Робертс К., Уотсон Дж. 1994
Введение в биологию клетки
Как изучают клетки?
Микроскопия
Клетки очень малы по размеру и сложно устроены: трудно рассмотреть их структуру, трудно определить молекулярный состав и еще труднее установить, как функционируют их отдельные элементы. Для изучения клеток разработано множество экспериментальных методов, возможности которых определяют уровень наших знаний в этой области, Успехи в изучении биологии клетки, включая наиболее удивительные достижения последних лет, как правило, связаны с применением новых методических подходов. Поэтому для понимания клеточной биологии необходимо иметь некоторое представление о соответствующих экспериментальных методах.
В этой главе мы вкратце рассмотрим современные методы, используемые для изучения клеток. Мы начнем знакомиться с теми из них, которые позволяют изучать клетку как единое целое, и затем обратима к анализу составляющих клетку макромолекул. Отправной точкой станет микроскопия, поскольку клеточная биология началась со световой микроскопии, и этот метод до сих пор остается весьма эффективным инструментом исследования, наряду с более современным устройствами для получения изображения, основанными на электронных пучках или иных формах излучения. От пассивного наблюдения мы постепенно перейдем к методам, предполагающим активное вмешательство: рассмотрим, как клетки различных типов могут быть отделены от ткани и при этом сохранять способность расти, узнаем, как клетки можно разрушить, а клеточные органеллы и составляющие их макромолекулы выделить в чистом виде. И наконец, мы изложим суть технологии рекомбинантных ДНК, благодаря которой стало возможным выделять, секвенировать и манипулировать генами и, следовательно, изучать механизмы их действия в клетке.
Диаметр типичной клетки животных составляет 10-20 мкм, что в пять раз меньше мельчайшей видимой частицы. Только с появлением совершенных световых микроскопов в начале XIX века удалось установить тот факт, что все ткани животных и растений состоят из отдельных клеток. Это открытие, обобщенное в форме клеточной теории Шлейденом и Шванном в 1838 году, знаменует собой начало клеточной биологии.
Будучи чрезвычайно малыми по размерам, животные клетки к тому же бесцветны и прозрачны; следовательно, открытие их основных структур стало возможным благодаря разработке набора красителей в конце XIX столетия. Именно красители обеспечили достаточный контраст для наблюдения субклеточных структур. Сходная ситуация наблюдалась в начале 40-х годов нашего столетия, когда изобретение мощного электронного микроскопа потребовало новых методов сохранения и окраски клеток. И только после того, как они были разработаны, начала проявляться вся сложность клеточной структуры, В основе микроскопии как методологии до сих пор лежат способы приготовления образца и возможности самого микроскопа.
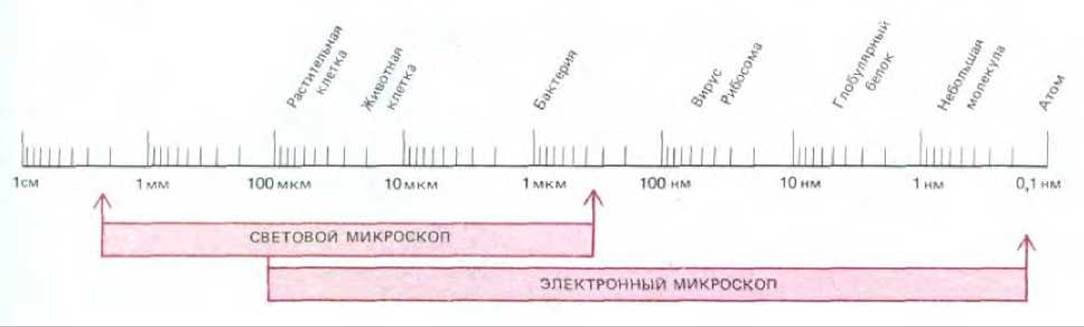
Рис. 4-1. Размеры клеток и клеточных компонентов, а также рабочие диапазоны светового и электронного микроскопа, изображенные в логарифмической шкале. В микроскопии принято пользоваться следующими единицами длины: мкм (микрометр) 10-6 м, нм (нанометр) - 10-9 м, A (ангстрем) - 10-10 м.
Таблица 4-1. Основные вехи в истории световой микроскопии
|
1611 - Кеплер (Kepler) предложил принцип создания сложного светового микроскопа 1655 - Гук (Hook) использовал сложный микроскоп для описания небольших пор в срезах пробки, названных им «клетками» 1674 - Левенгук (Leeuwenhoek) сообщил об открытии им одноклеточных. Спустя 9 лет он впервые увидел бактерии 1833 - Браун (Brown) опубликовал свои микроскопические наблюдения над орхидеями, в которых он четко описал ядро клетки 1838 - Шлейден и Шванн (Schleiden, Schwann) предложили клеточную теорию, согласно которой структурной и функциональной единицей строения растений и животных является клетка, содержащая ядро 1857 - Колликер (Kolliker) описал митохондрии в мышечных клетках 1876 - Аббе (Abbe) проанализировал влияние дифракции на формирование изображения и показал возможность усовершенствования конструкции микроскопа 1879 - Флемминг (Flemming) с большой точностью описал поведение хромосом во время митоза у животных клеток 1881 - Ретциус (Retzius) наиболее подробно описал многие ткани животных. В течение следующих 20 лет он, Кахал (Cajal) и другие гистологи разработали методы окрашивания тканей и заложили основы микроскопической анатомии 1882 - Кох (Koch) для окрашивания микроорганизмов использовал анилиновые красители и идентифицировал бактерии, вызывающие туберкулез и холеру. В течение последующих 20 лет другие бактериологи, в том числе Клебс и Пастер (Klebs, Pasteur), выявили и описали возбудителей многих болезней, изучая окрашенные препараты под микроскопом 1886 - Цейсе (Zeiss), используя идею Аббе (Abbe), изготовил серию линз. Благодаря этому усовершенствованию, микроскописты смогли различать структуры, размеры которых были соизмеримы с теоретическим пределом разрешения для видимого света 1898 - Гольджи (Golgi), окрашивая клетки азотнокислым серебром, впервые наблюдал и описал аппарат Гольджи 1924 - Лакассань (Lacassagne) и его сотрудники разработали первые методы радиоавтографии для выявления радиоактивного полония в биологических образцах 1930 - Лебедев разработал и создал первый интерференционный микроскоп. В 1932 г. Зернике (Zernicke) изобрел фазово-контрастный микроскоп. Эти два изобретения позволили наблюдать неокрашенные живые клетки и изучать их строение 1941 - Кунс (Coons) для выявления клеточных антигенов использовал антитела, связанные с флуоресцирующими красителями 1952 - Номарский (Nomarski) разработал и запатентовал систему дифференциального интерференционного контраста для светового микроскопа, которая до сих пор носит его имя |

Рис. 4-2. Интерференция световых волн. Если две световые волны совпадают по фазе, амплитуда результирующей волны возрастает и, следовательно, яркость увеличивается. Если две световые волны не совпадают по фазе, они гасят друг друга и образуют волну, амплитуда которой (а следовательно, и яркость) уменьшается.

Рис. 4-3. Эффект интерференции, который можно наблюдать при большом увеличении по краям твердого объекта, расположенного между источником света и наблюдателем.
На рис. 4-1 сравниваются степени разрешения в современном световом и электронном микроскопах.
Основные этапы развития современной микроскопии перечислены в табл. 4-1.
4.1.1. С помощью светового микроскопа можно различить объекты, отстоящие друг от друга на 0,2 мкм [2]
В общем случае излучение данной длины волны может быть использовано для изучения только таких структур, минимальные размеры которых еще сопоставимы с длиной волны самого излучения. Этот фундаментальный принцип ограничивает возможности любого микроскопа. Предел разрешения светового микроскопа задается длиной световой волны, которая для видимого света лежит в пределах от 0,4 мкм (фиолетовый) до 0,7 мкм (темно-красный). Из этого следует, что самыми маленькими объектами, которые еще можно наблюдать в световой микроскоп, являются бактерии и митохондрии (их ширина ~0,5 мкм). Более мелкие элементы клетки искажаются эффектами, вызванными волновой природой света. Чтобы понять природу этих эффектов, мы должны проследить за тем, что происходит со световыми волнами по мере их прохождения сквозь линзы микроскопа.
Вследствие волновой природы света его луч не движется по идеально прямому пути, предсказываемому законами геометрической оптики. В реальной ситуации световые волны перемещаются сквозь оптическую систему по множеству слегка отличающихся путей. Оптическая дифракция обусловлена интерференцией световых волн, пути прохождения которых через оптическую систему несколько различаются. Если световые волны точно совпадают по фазе, т. е. гребень одной соответствует гребню другой, а впадина одной - впадине другой, то они взаимно усиливаются, и яркость возрастает. С другой стороны, если фазы волн не совпадают, они будут взаимно погашаться (рис. 4-2). Тень прямого края, например, освещенного светом одной длины волны, при большом увеличении будет выглядеть как набор параллельных линий, тогда как округлое пятно проявится в виде набора концентрических окружностей (рис. 4-3). По этой же причине отдельная точка выглядит в микроскопе, как яркое пятно, а два ближайших точечных объекта дают перекрывающиеся изображения, которые сливаются в одно. Повышение точности обработки линз не позволяет преодолеть это ограничение, поскольку оно задано волновой природой света.
Предельное разрешение, при котором два объекта могут наблюдаться в отдельности, так называемый предел разрешения - зависит как от волновой природы света, так и от апертуры использованной системы линз (рис. 4-4). В наиболее благоприятных условиях - при фиолетовом свете (длина волны = 0,4 мкм) и апертуре 1,4 можно достигнуть теоретически возможного предела разрешения светового микроскопа около 0,2 мкм. Этот предел был достигнут конструкторами микроскопов в конце XIX столетия (однако в современных микроскопах, производимых серийно, достигается очень редко). И хотя изображение можно увеличить как угодно, например, проецируя его на экран, все же в световой микроскоп нельзя разрешить два объекта, если они разделены расстоянием менее 0,2 мкм: такие объекты будут выглядеть как один объект.
Волновая природа света не всегда является помехой в изучении клеток, позже мы увидим как интерференция и дифракция могут быть использованы для изучения живых неокрашенных клеток. Но сначала необходимо обсудить методы получения постоянных препаратов клеток и то, как с помощью химических красителей можно улучшить возможности наблюдения клеточных структур в таких препаратах.

Рис. 4-4. Направление движения световых волн, проходящих сквозь прозрачный образец в микроскопе. Иллюстрирует концепцию апертуры и ее связь с ограничением разрешения.
4.1.2. Для проведения микроскопических исследований ткани обычно фиксируют и режут [2]
Для приготовления постоянного препарата, который можно окрасить и наблюдать в микроскоп, необходимо сначала обработать клетки фиксирующим агентом с тем, чтобы иммобилизировать, убить и сохранить их. Используя химические термины можно сказать, что фиксация повышает доступность клеток красителям; макромолекулы клеток скрепляются поперечными сшивками, что стабилизирует и закрепляет их в определенном положении. Некоторые ранние методы фиксации включали обработку кислотами или органическими растворителями, например, спиртом. В современных методах, как правило, используется обработка альдегидами, например, формальдегидом или глутаральдегидом, которые формируют ковалентные связи со свободными аминогруппами белков и, таким образом, сшивают соседние молекулы.
Толщина большинства образцов тканей слишком велика, чтобы можно было непосредственно изучать отдельные клетки при высоком разрешении. Поэтому после фиксации ткани обычно режут на очень тонкие «ломтики» (срезы) на микротоме: это прибор с очень острым металлическим лезвием, который действует подобно хлеборезке (рис. 4-5). Срезы толщиной от 1 до 10 мкм помещают на поверхность предметного стекла. Обычно ткани очень мягки и нежны даже после фиксации и их необходимо перед разделением на срезы заключать в поддерживающую среду. Как правило, в качестве заключающих сред используют парафин или специальную смолу. В жидком виде эти среды пропитывают и окружают фиксированную ткань.

Рис. 4-5. Приготовление среза на микротоме после заливки ткани. Срез предназначен для исследования с помощью светового микроскопа. девают при охлаждении или за счет полимеризации, образуя твердый блок, который удобно резать на микротоме.
Существует серьезная опасность того, что процедуры фиксации или заключения могут повредить структуру клеток или клеточных макромолекул. Вот почему предложен другой метод приготовления срезов, уменьшающий эту опасность, - быстрое замораживание. Здесь можно обойтись без фиксации и заливки. Замороженную ткань просто режут на криостате - специальном микротоме, установленном в холодной камере. Полученные таким образом срезы позволяют избежать некоторых артефактов, и в то же время обладают определенными недостатками: отдельные структуры индивидуальных макромолекул, таких, например, как белки, при замораживании сохраняются хорошо, но структура самой клетки может оказаться поврежденной. Следующий этап после приготовления срезов (любым из методов) - их окраска.
4.1.3. Различные компоненты клетки можно окрашивать по-разному [3]
В содержимом большинства клеток, состоящих, как правило, на 70% из воды, практически отсутствуют компоненты, способные помешать прохождению световых лучей. Поэтому в естественном состоянии большинство клеток даже после фиксации и приготовления срезов практически невидимы в обычном световом микроскопе. Одна из возможностей их увидеть состоит в окраске клеток красителями.
В начале XIX столетия ввиду потребности в красителях для текстильной промышленности, органическая химия переживала очень плодотворный период. Оказалось, что некоторые из этих красителей также способны окрашивать биологические ткани. К удивлению исследователей, некоторые из этих красителей обладали определенным сродством к специфическим компонентам клетки - ядру или митохондриям, окрашивая их внутренние структуры и делая их доступными для изучения под микроскопом. В настоящее время известен широкий набор органических красителей. Многие из них обладают весьма колоритными названиями: малахитовый зеленый, судан черный, кумасси голубой; каждый краситель характеризуется сродством к определенным субклеточным компонентам. Например, краситель гематоксилин имеет сродство к отрицательно заряженным молекулам и поэтому выявляет распределение в клетках ДНК и кислых белков. Химическая природа специфичности многих красителей до сих пор неизвестна.
Накопление опыта в клеточной химии сопровождалось отбором наиболее рациональных и избирательных методов окраски и, в частности, методов, позволяющих отличать специфические белки либо иные макромолекулы клеток. Здесь же возникла проблема чувствительности. Поскольку большинство макромолекул представлены в клетках относительно незначительным числом копий, одна или две молекулы красителя, связанные с макромолекулой, могут оставаться незамеченными. Один из путей разрешения этой проблемы состоял в увеличении числа молекул красителя, ассоциированных с отдельными клеточными молекулами. По каталитической активности в клетках удалось локализовать многие ферменты: при достаточном обеспечении соответствующим субстратом каждая молекула фермента создавала множество молекул видимого продукта реакции. Альтернативный подход к проблеме чувствительности состоит в использовании флуоресценции. В данном случае можно на темном фоне выявлять специфические красители по свету, который они и только они излучают, будучи соответствующим образом возбуждены. Далее мы переходим к объяснению этого феномена.
4.1.4. Специфические молекулы могут быть локализованы в клетках с помощью флуоресцентной микроскопии [4]
Флуоресцирующие красители поглощают свет одной длины волны и излучают свет другой длины волны, более длинной. Если такое вещество облучить светом, длина волны которого совпадает с длиной волны света, поглощаемого красителем, и затем для анализа использовать фильтр, пропускающий свет с длиной волны, соответствующей свету, излучаемому красителем, флуоресцирующую молекулу можно выявить по свечению на темном поле. Высокая интенсивность излучаемого света является характерной особенностью таких молекул. Применение флуоресцирующих красителей для окраски клеток предполагает использование специального флуоресцентного микроскопа. Такой микроскоп похож на обычный световой микроскоп, но здесь свет от осветителя, излучаемый мощным источником, проходит через два набора фильтров - один для задержания света перед образцом и другой для фильтрации света, полученного от образца. Первый фильтр выбран таким образом, что он пропускает свет длины волны, возбуждающей определенный флуоресцирующий краситель; в то же время второй фильтр блокирует этот падающий свет и пропускает на окуляр свет длины волны, излучаемой красителем при его флуоресценции (рис. 4-6). Флуоресцентная микроскопия часто используется для выявления специфических белков или других молекул, которые становятся флуоресцирующими после ковалентного связывания с флуоресцирующими красителями. Например, флуоресцирующие красители могут быть связаны с молекулами антител, что сразу же превращает их в высокоспецифические и удобные красящие реагенты, селективно связывающиеся со специфическими макромолекулами на поверхности живой либо внутри фиксированной клетки (см. разд. 4.5.3). Для этой цели обычно используют два красителя - флуоресцеин, который дает интенсивную желто-зеленую флуоресценцию после возбуждения светло-голубым светом, и родамин, обусловливающий темно-красную флуоресценцию после возбуждения желто-зеленым светом (рис. 4-7). Применяя для окраски флуоресцеин и родамин, можно изучать распределение различных молекул, два вида молекул будут выявляться в микроскопе после простого переключения двух наборов фильтров, каждый из которых специфичен для одного из красителей (рис. 4-8).
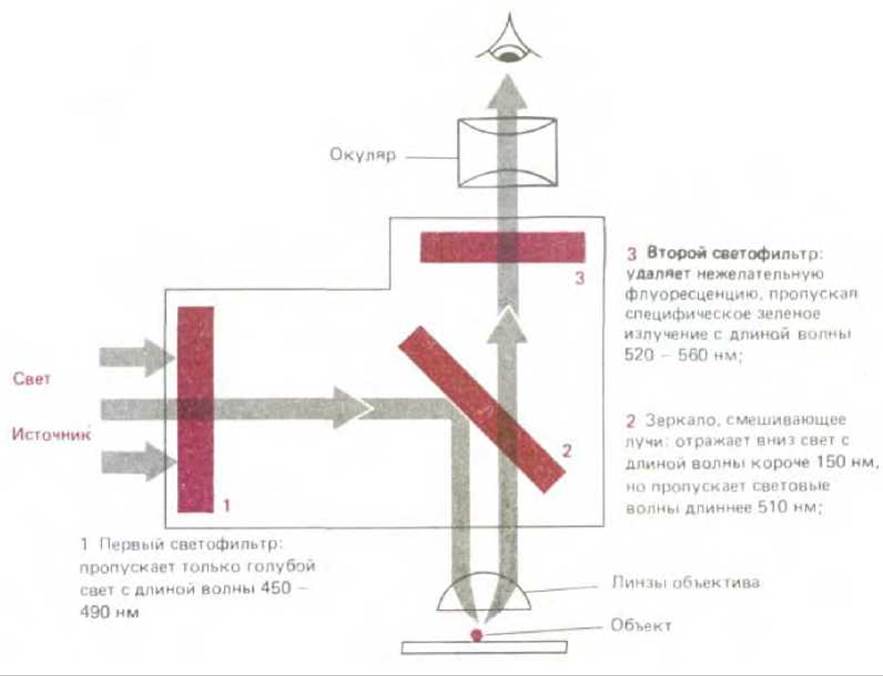
Рис. 4-6. Оптическая система современного флуоресцентного микроскопа состоит из двух избирательных фильтров и дихроического (смешивающего лучи) зеркала. Здесь указан набор фильтров, используемых для выявления свечения флуоресцеина. Для микроскопа этого типа важно наличие в объективе линз, характеризуемых высокой апертурой, так как для данного увеличения яркость изображения пропорциональна одной четвертой апертуры (см. также рис. 4-4).

Рис. 4-7. В флуоресцентной микроскопии обычно используют два красителя флуоресцеин и тетраметилродамин, структуры которых представлены на данном рисунке. Флуоресцеин излучает желто-зеленый свет после активации светом соответствующей длины волны. Родамин излучает красный свет. Часть молекулы, обозначенная цветом, указывает на расположение химически активной группы; в этом положении, как правило, формируется ковалентная связь между красителем и белком (или иной молекулой). В настоящее время промышленность выпускает несколько вариантов этих красителей с различными типами реакционно'-активных групп, что позволяет «нацелить» этот краситель либо на SH-группы, либо на NН2-группы белка.
Далее мы переходим к обсуждению новых важных методов, которые позволяют использовать флуоресцентную микроскопию для анализа изменений концентрации и расположения специфических макромолекул в живых клетках (разд. 4.1.9).
4.1.5. Фазово-контрастный и интерференционный микроскопы позволяют изучать живые клетки[2]
Возможность потери или нарушения образцов в процессе их приготовления всегда беспокоила микроскопистов. Единственный способ решить эту проблему состоит в изучении живых клеток без фиксации или замораживания. Для этой цели очень полезны микроскопы со специальными оптическими системами.
При прохождении света через живую клетку фаза световой волны меняется согласно коэффициенту рефракции клетки: свет, проходящий через относительно тонкие или относительно толстые участки клетки, такие, как ядро, задерживается, и его фаза соответственно сдвигается по отношению к фазе света, проходящего через относительно тонкие участки цитоплазмы. Как в фазово-контрастном, так и в интерференционном микроскопе используются эффекты интерференции, возникающие при рекомбинации двух наборов волн, которые и создают изображение клеточных структур (рис. 4-9). Оба типа световой микроскопии широко используются для наблюдения живых клеток.

Рис. 4-8. Флуоресцентная микрофотография участка поверхности ранних эмбрионов Drosophila, микротрубочки которых были помечены антителами, связанными с флуоресцеином (слева), а актиновые филаменты - антителами, меченными родамином (в центре). Кроме того, хромосомы были помечены третьим красителем, который флуоресцирует, только связавшись с ДНК (справа). На этой стадии все ядра эмбриона расположены в общей цитоплазме и не разделены клеточными стенками; они находятся в метафазе митоза. Все три микроснимка были сделаны с одного участка фиксированного эмбриона с использованием трех различных наборов фильтров в флуоресцентном микроскопе (см. также рис. 4-6). (С любезного разрешения Tim Carr).

Рис. 4-9. Два способа увеличения контраста в световой микроскопии. А. Окрашенные участки клетки уменьшают амплитуду проходящих световых волн определенной длины. В результате можно получить окрашенное изображение, видимое при прямом наблюдении. Б. Амплитуда световых волн, проходящих через неокрашенную живую клетку, практически не меняется; поэтому многие детали нельзя увидеть при прямом наблюдении. Здесь, однако, имеет место изменение фазы проходящего света явление, используемое в фазово-контрастном и интерференционном микроскопах для получения высококонтрастного изображения.
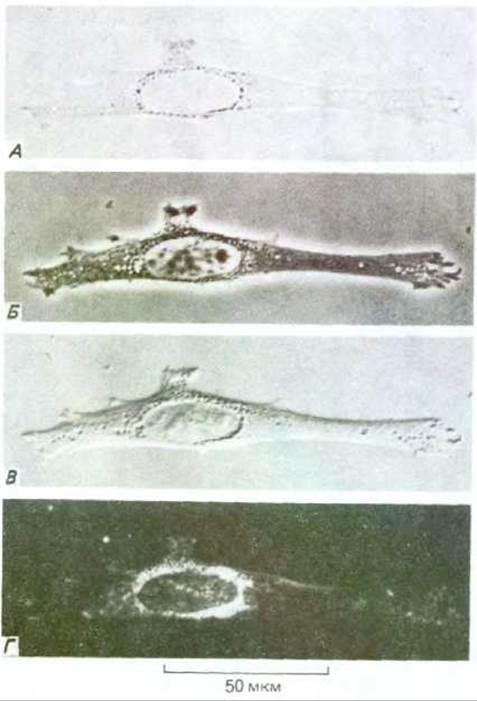
Рис. 4-10. Фибробласт в культуре ткани при наблюдении с помощью четырех различных типов световой микроскопии. А. Изображение получено при прямом прохождении лучей через клетку (микроскопия в светлом поле). Остальные изображения получены с помощью методов, рассматриваемых в тексте: Б-фазово-контрастная микроскопия; В -интерференционная микроскопия; Г микроскопия в темном поле. Простая замена компонентов оптики большинства современных микроскопов позволяет получать все четыре типа изображения.

Рис. 4-11. Изображение неокрашенных микротрубочек, наблюдаемое в интерференционном микроскопе. А. Исходное необработанное изображение; Б. Изображение, полученное после электронной обработки, существенно увеличивающей контраст и снижающей «шум». Хотя диаметр микротрубочек всего 0,025 мкм, вследствие дифракции они выглядят, как значительно более толстые филаменты. (С любезного разрешения Вruсе Sohnapp.)
Простейший способ разглядеть детали клеточной структуры - наблюдать свет, рассеивающийся различными компонентами клетки. В темнопольном микроскопе лучи от осветителя направляются сбоку и при этом в линзы микроскопа попадают только рассеянные лучи. Соответственно клетка выглядит как освещенный объект на темном поле. Изображение одной и той же клетки, полученное четырьмя способами световой микроскопии, показано на рис. 4-10.
Одним из основных преимуществ фазово-контрастной, интерференционной и темнопольной микроскопии является возможность наблюдать движение клеток в процессе митоза и миграции. Клеточные движения, как правило, совершаются очень медленно и их сложно наблюдать в реальном времени. В этом случае используют покадровую (цейтраферную) микрокиносъемку или видеозапись. Последовательные кадры при этом разделены во времени, но при воспроизведении записи с нормальной скоростью картина реальных событий ускоряется.
4.1.6. Изображение можно усиливать или анализировать с помощью электронных методов [5]
В последние годы видеокамеры и соответствующие технологии обработки изображения значительно увеличили возможности световой микроскопии. Благодаря их применению удалось не только преодолеть трудности, связанные с несовершенством оптической системы, но и решить проблемы, обусловленные особенностями физиологии человека. Они состоят в том, что:
1) глаз не регистрирует очень слабый свет;
2) глаз не способен фиксировать небольшие отличия в интенсивности света на ярком фоне.
Первая из этих проблем была преодолена после присоединения к микроскопу сверхвысокочувствительных видеокамер (подобных тем, которые используются при ночных съемках). Это позволило наблюдать клетки в течение длительного времени при низкой освещенности, исключая длительное воздействие яркого света (или тепла). Системы усиления изображения особенно важны для изучения в живых клетках флуоресцирующих молекул.
Поскольку изображение создается видеокамерой в форме электронных сигналов, его можно соответствующим образом преобразовать в числовые сигналы, направить в компьютер и затем подвергнуть дополнительной обработке для извлечения скрытой информации. Эти и подобные методы обработки изображения позволяют компенсировать оптические недостатки микроскопов и практически достичь предела разрешения. Более того, используя современные видеосистемы, контраст может быть усилен настолько, что преодолеваются ограничения глаза в детектировании небольших отклонений интенсивности света. Хотя этот процесс усиливает случайные отклонения фона в оптической системе, такой «шум» может быть устранен специальными методами. Таким образом, благодаря современным подходам мы получили возможность анализировать прозрачные объекты, ранее не отличимые от фона.
Высокий контраст, достижимый с помощью компьютерной интерференционной микроскопии, позволяет наблюдать даже очень мелкие объекты, как, например, отдельные микротрубочки (рис. 4-11), диаметр которых менее одной десятой длины волны света (0,025 мкм). Отдельные микротрубочки можно увидеть и с помощью флуоресцентной микроскопии (см. рис. 4-56). Однако в обоих случаях неизбежны эффекты дифракции, сильно изменяющие изображение. Диаметр микротрубочек при этом завышается (0,2 мкм), что не позволяет отличать отдельные микротрубочки от пучка из нескольких микротрубочек. Для решения этой задачи необходим электронный микроскоп, предел разрешения которого сдвинут далеко за пределы длины волны видимого света.

Рис. 4-12. Электронная микроскопия тонкого слоя золота позволяет выявить отдельные атомы в виде отдельных ярких пятен. Расстояние между соседними атомами золота около 0,2 нм. (С любезного разрешения Graham Hills.)
4.1.7. Электронный микроскоп позволяет анализировать тонкую структуру клетки [6]
Взаимосвязь длины волны света и предела разрешения (см. рис. 4-4) сохраняется для любой формы излучения, как для световых лучей, так и для электронов. Однако в последнем случае предел разрешения существенно ниже. Длина волны электрона уменьшается с увеличением его скорости. В электронном микроскопе с напряжением 100000 В длина волны электрона равна 0,004 нм, а согласно теории, разрешение такого микроскопа составляет 0,002 нм. Однако коррекция аберрации электронных линз - задача более сложная, чем для стеклянных линз, и поэтому в реальности разрешение современных электронных микроскопов в лучшем случае 0,1 нм (1 А) (рис. 4-12). Более того, трудности приготовления образца, его повреждение излучением существенно снижают нормальное разрешение, которое для биологических объектов составляет 2 нм (20 А) (т.е. примерно в 100 раз выше, чем у светового микроскопа).
Некоторые из достижений в развитии электронной микроскопии перечислены в табл. 4-2.
Общая схема просвечивающего электронного микроскопа (ПЭМ) напоминает схему светового, хотя электронный микроскоп значительно больше и как бы перевернут (рис. 4-13). Источник излучения - нить катода, испускающая электроны с вершины цилиндрической колонны высотой около двух метров. Поскольку при столкновении с молекулами воздуха электроны рассеиваются, в колонне должен быть создан вакуум. Электроны, излучаемые катодной нитью, ускоряются ближайшим анодом и проникают через крошечное отверстие, формируя электронный луч, проходящий в нижнюю часть колонны. Вдоль колонны на некотором расстоянии расположены кольцевые магниты, фокусирующие электронный луч, подобно стеклянным линзам, фокусирующим луч света в световом микроскопе. Образец через воздушный шлюз помещают в вакуум колонны, на пути электронного пучка. Часть электронов в момент прохождения через образец рассеивается согласно плотности вещества в данном участке, остаток электронов фокусируется и образует изображение (подобно формированию изображения в световом микроскопе) на фотопластинке или на фосфоресцирующем экране.
4.1.8. Для наблюдения под электронным микроскопом биологические образцы необходимо подвергнуть специальной обработке [7]
Применение электронного микроскопа в биологии позволило увидеть в клетках множество удивительных структур. Но прежде, чем эти открытия были сделаны, ученым пришлось изрядно потрудиться, чтобы разработать новые методы заключения тканей, их резки и окрашивания.
Таблица 4-2. Основные вехи в истории электронной микроскопии
|
1897 - Томсон (J. J. Thomson) сообщил о существовании отрицательно заряженных частиц, названных позже электронами 1924 - Де Бройль (de Broglie) предположил, что движущийся электрон обладает волновыми свойствами 1926 - Буш (Busch) доказал, что с помощью цилиндрических магнитных линз можно сфокусировать электронный луч. Так были заложены основы электронной оптики 1931 - Руска (Ruska) и соавторы создали первый просвечивающий электронный микроскоп 1935 - Кнолль (Knoll) показал возможность создания сканирующего электронного микроскопа; спустя три года фон Ардене (Von Ardene) конструировал его прототип 1939 - Сименс (Siemens) создал первый просвечивающий электронный микроскоп, который нашел широкое применение. 1944 - Уильяме и Виков (Williams, Wyckoff) разработали метод оттенения металлом 1945 - Портер, Клод и Фуллам (Porter, Claude, Fullam) использовали электронный микроскоп для изучения клеток в культурах тканей после их фиксации и окраски 1948 - Пиз и Бейкер (Pease, Baker) представили убедительные доказательства, что ими получены тонкие срезы биологического материала (толщиной 0,1-0,2 мкм) 1952 - Паладе, Портер и Шестранд (Palade, Porter, Sjostrand) разработали методы фиксации и приготовления тонких срезов. Это позволило впервые увидеть многие внутриклеточные структуры. Хаксли (Н. Е. Xuxley) был одним из первых, кто применил эти методы, и ему удалось показать, что скелетная мышца содержит перекрывающиеся сети белковых филаментов. Так были получены доказательства в пользу гипотезы «скользящих нитей», объясняющей сокращение мышцы 1953 - Портер и Блюм (Porter, Blum) разработали ультрамикротом, который первым нашел широкое применение. В нем были использованы многие принципы, предложенные ранее Клодом и Шестрандом (Claude, Sjostrand) 1956 - Глауэрт (Glauert) и сотрудники показали, что смола аралдит является высокоэффективным средством для заключения препаратов в электронной микроскопии. Пятью годами позже Люфт (Luft) предложил другую смолу для заключения - эпон 1957 - Робертсон (Robertson) первым наблюдал в электронный микроскоп и описал трехслойное строение клеточной мембраны 1957 - Метод «замораживание-скалывание», разработанный Стиром (Steere), был усовершенствован Муром и Мюреталером (Moor, Muhlethaler). Позже (в 1966 г.), Брентон (Branton) показал, что данный метод позволяет изучать внутреннее строение мембран 1959 - Бреннер и Хорн (Bretftier, Home) усовершенствовали метод негативного контрастирования, который был разработан Холлом (Hall) четырьмя годами ранее, после чего этот метод вошел в широкую практику 1959 - Сингер (Singer) использовал антитела, связанные с ферритином, для выявления молекул клетки с помощью электронной микроскопии 1963 - Сабатини, Бенш и Баррнетт (Sabatini, Bensch, Barrnett) начали использовать глутаральдегид (с последующей обработкой OsO4) в качестве фиксатора для электронной микроскопии 1965 - Фирма «Кембридж Инструменте» (Cambridge Instruments) впервые выпустила для продажи сканирующий электронный микроскоп 1968 - Де Розьер и Клуг (de Rosier, Klug) описали метод определения трехмерных структур по электронным микрофотографиям 1975 - Хендерсон и Унвин (Henderson, Unwin) впервые определили тонкое строение мембранного белка, используя реконструкцию электронных микрофотографий неокрашенных белков на компьютере 1979 - Хейзер, Рис (Heuser, Reese) и сотрудники разработали высокоразрешающий метод глубокого травления, основанный на сверхбыстром замораживании |
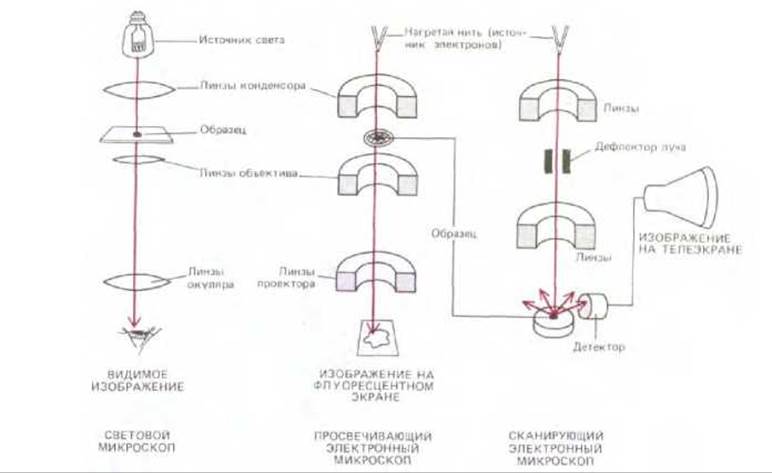
Рис. 4-13. Схематическое изображение основных узлов светового микроскопа, а также просвечивающего и сканирующего электронных микроскопов, подчеркивающее сходные черты в конструкции этих приборов. В электронных микроскопах обоих типов образец должен быть помещен в вакуум.

Рис. 4-14. Глутаральдегид и тетраоксид осмия наиболее распространенные фиксаторы, используемые в электронной микроскопии. Две реакционноспособные альдегидные группы глутаральдегида позволяют сшивать различные типы молекул с помощью ковалентных связей, вытесняющих атомы водорода (выделены цветом). Тетраоксид осмия восстанавливает многие органические соединения, с которыми образует поперечно-сшитые комплексы. Это свойство особенно ценно в случае клеточных мембран, поскольку тетраоксид осмия реагирует с двойными связями С -С, характерными для многих жирных кислот.
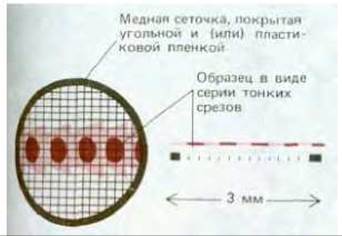
Рис. 4-15. Схематическое изображение медной сеточки, используемой для поддержания тонких срезов образца в просвечивающем электронном микроскопе.
В электронном микроскопе все образцы подвергаются действию вакуума высокой степени разрежения и поэтому их невозможно наблюдать в живом влажном состоянии. Ткани обычно сохраняют с помощью фиксации - сначала глутаральдегидом, ковалентно связывающим белковые молекулы между собой, а затем осмиевой кислотой, связывающей и стабилизирующей двойной слой липидов и белки (рис. 4-14). Электроны обладают низкой проникающей способностью, этим вызвана необходимость получения срезов толщиной от 50 до 100 нм (1/200 толщины одной клетки). Только такие срезы можно наблюдать в электронный микроскоп. Для этого потребовалось также предварительное обезвоживание образца и пропитка мономерными смолами, которые после полимеризации создают твердый блок пластмассы, заключающий образец; блок затем режут с помощью тонкого стеклянного или алмазного ножа на специальном микротоме. Полученные срезы не содержат воды или иных летучих органических веществ; такие срезы помещают на небольшую круглую металлическую сетку для наблюдения под микроскопом (рис. 4-15).
Контраст, создаваемый в электронном микроскопе, определяется атомным числом веществ образца. Чем выше атомное число, тем больше электронов рассеивается и тем выше контраст. В состав биологических молекул входят атомы с очень низким атомным числом (в основном кислород, водород, углерод и азот). Для усиления контраста образцы до и после резки импрегнируют солями тяжелых металлов, таких, как осмий, уран и свинец. Компоненты клетки выявляются с разной степенью контраста согласно степени их импрегнации (или окраски) этими солями. Как правило, липиды окрашиваются осмием в темный цвет и это позволяет выявлять мембраны (рис. 4-16).
С помощью методов электронной микроскопии в некоторых случаях на тонких срезах удается локализовать специфические макромолекулы. Некоторые ферменты клеток выявляются после инкубации образцов с субстратами, ферментативная реакция с которыми приводит к местному отложению электроноплотного осадка (рис. 4-17). Кроме того, для локализации специфических макромолекул можно использовать меченые антитела, если их связать с индикаторным ферментом пероксидазой или электроноплотным маркером, например, частицами коллоидного золота (см. разд. 4.5.3).

Рис. 4-16. Тонкий срез верхушки корня злака. Легко различимы клеточная стенка, ядро, вакуоли, митохондрии, эндоплазматический ретикулум, аппарат Гольджи и рибосомы. (С любезного разрешения Brian Gunning.)

Рис. 4-17. Электронная микрофотография клетки, иллюстрирующая локализацию конкретного фермента (нуклеотиддифосфатазы) в аппарате Гольджи. Тонкий срез клетки инкубирован с субстратом, образующим под действием фермента электроноплотный осадок. (С любезного разрешения Daniel Friend.)

Рис. 4-18. Схематическое изображение, иллюстрирующее, как легко прийти к ошибочным выводам, изучая отдельные тонкие срезы. Так, например, в данном случае рассматриваются срезы клетки, в которой имеется лишь одна разветвленная митохондрия. Между тем создастся впечатление, что большинство срезов содержит две или три отдельные митохондрии. Более того, может показаться, что на срезах 4 и 7 выявляемая митохондрия находится в процессе деления. Серийные срезы позволяют реконструировать реальную форму, существующую в действительности.
4.1.9. Сканирующий электронный микроскоп используется для получения трехмерного изображения поверхности [8]
Тонкие срезы практически являются двумерными срезами ткани и не позволяют судить о трехмерной структуре клеточных компонентов. Трехмерное изображение можно получить после реконструкции сотен серийных срезов (рис. 4-18), но это долгий и утомительный процесс. В настоящее время разработаны более прямые методы получения трехмерного изображения. Один из них состоит в изучении образца под сканирующим электронным микроскопом (СЭМ), который обычно меньше и проще, чем просвечивающий электронный микроскоп. Для получения изображения в просвечивающем электронном микроскопе используют электроны, проходящие через образец, а в сканирующем электронном микроскопе используются электроны, рассеиваемые или излучаемые поверхностью образца. В данном случае образец должен быть зафиксирован, высушен и покрыт тонкой пленкой тяжелого металла. Затем образец сканируется очень узким пучком электронов. При этом оценивают количество электронов, рассеиваемых при облучении последовательных точек металлической поверхности. Полученное значение используют для контроля интенсивности второго луча, движущегося синхронно первому и формирующему изображение на телевизионном экране. Таким образом происходит формирование единого, цельного и значительно увеличенного изображения.

Рис. 4-19. Полученная с помощью сканирующего электронного микроскопа микрофотография стереоцилий, расположенных в виде органных труб на поверхности волосковых клеток внутреннего уха. (С любезного разрешения R. Jackobs, A.J. Hudspeth.)

Рис. 4-20. Электронная микрофотография отдельных молекул белка миозина (оттенение платиной). Миозин основной компонент сократительного аппарата мышц; здесь можно увидеть, что он состоит из двух глобулярных участков, связанных с общим палочковидным хвостом. (С любезного разрешения Arthur Elliot.)
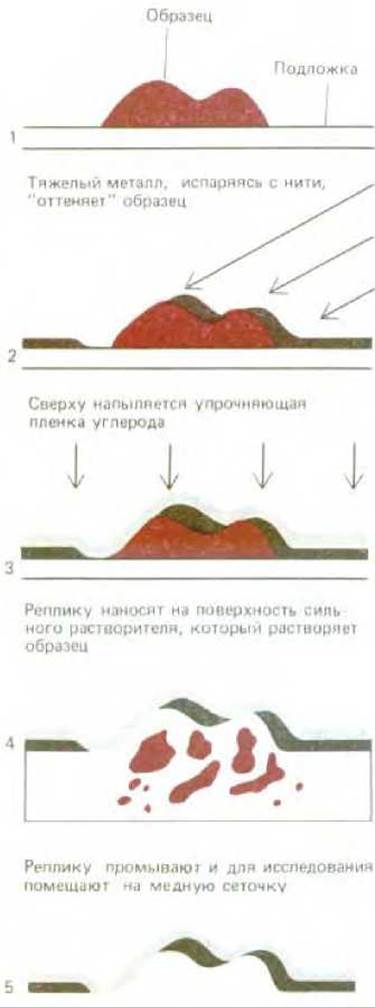
Рис. 4-21. Схематически показан метод приготовления реплики с поверхности образца (оттенение металлом). Обратите внимание, что толщина слоя металла определяется контуром поверхности исходного образца.
Метод сканирующей электронной микроскопии обеспечивает значительную глубину фокусировки; более того, поскольку масштабы рассеивания электронов определяются углом поверхности по отношению к лучу, на изображении возникают чередующиеся светлые и темные участки, создающие впечатление трехмерности (рис. 4-19). Но этот метод применим только для изучения поверхности и его разрешение сравнительно невелико (около 10 нм с эффективным увеличением примерно 20 тыс. раз). Данный метод практически неприменим для изучения субклеточных органелл и используется исключительно для изучения целых клеток и тканей.
Используя обычные тонкие срезы, поворачивая их и фотографируя их под разными углами, можно и в обычном просвечивающем электронном микроскопе получить имитацию трехмерного изображения. При наблюдении в стереоочки полученной стереопары изображений объекта возникает псевдотрехмерное изображение. Толщина образцов, изучаемых этим методом, определяется проникающей способностью электронов или их энергией. Совершенствование метода привело к созданию высоковольтных микроскопов с ускоряющим напряжением до 1 млн. вольт (против 100 тыс. вольт у обычных ПЭМ). Эти гигантские приборы позволяют изучать по методу просвечивающей электронной микроскопии срезы толщиной в несколько микрометров.
4.1.10. Для изучения деталей поверхности в просвечивающем электронном микроскопе используют оттенение [9]
Просвечивающий электронный микроскоп можно использовать для изучения поверхности образца с очень большим увеличением, наблюдая отдельные макромолекулы. Как и при сканирующей электронной микроскопии, на высушенный образец напыляется тонкая пленка тяжелого металла, например, платина. Металл напыляется под определенным углом, так что отложения напыленной пленки в некоторых местах толще, чем в других. Этот процесс известен как оттенение здесь возникает эффект тени, создающий впечатление трехмерности изображения.
Приготовленные таким образом образцы могут быть достаточно малы и тонки, чтобы электронный луч проникал сквозь них; например, таким способом можно анализировать индивидуальные молекулы. вирусы и стенки клеток (рис. 420). Что же касается более толстых образцов, то здесь после оттенения необходимо удалить органический материал клетки, при этом на поверхности образца останется только тонкий металлический отпечаток или реплика поверхности. Эта реплика затем усиливается углеродной пленкой, после чего ее можно поместить на сетку и изучать в обычном электронном микроскопе (рис. 4-21).
4.1.11. Методы электронной микроскопии замораживание-скалывание и замораживание травление обеспечивают уникальную возможность наблюдать внутреннее строение клетки [10]
В клеточной биологии особенно успешно используются два метода, основанные на получении механических реплик. Один из них - метод электронной микроскопии «замораживание-скалывание» - дает возможность изучать внутреннее строение клеточных мембран. Клетки замораживают при температуре жидкого азота (-196°С) в присутствии криопротектора (антифриза) во избежание искажений за счет образования кристаллов льда. Замороженный блок затем раскалывают лезвием ножа. Скол часто проходит через гидрофобную середину двойного слоя липидов, обнажая внутреннюю поверхность клеточных мембран. Образующуюся поверхность скола оттеняют платиной, органический материал удаляют и изучают полученные реплики в электронном микроскопе (рис. 4-21). Такие реплики усеяны небольшими выпячиваниями - внутримембранными частицами, которые представляют собой крупные мембранные белки. Этот метод чрезвычайно удобен и эффективен при анализе распределения таких белков на поверхности мембраны (рис. 4-22).

Рис. 4-22. Электронная микрофотография тилакоидной мембраны хлоропластов клетки растений, полученная по методу замораживания-скалывания. Тилакоидные мембраны, осуществляющие фотосинтез, уложены в виде множества слоев. Плоскость скола переходит с одного слоя на другой и проходит через середину каждого липидного бислоя. Внутримембранные белки, которые содержатся в значительном количестве внутри двойного слоя, обнажаются и после натенения выявляются в виде внутримембранных частиц в этой платиновой реплике. Самой крупной частицей, выявляемой на мембране, является комплекс из множества белков, образующих фотосистему II. (С любезного разрешения L. F. Staehelin.)
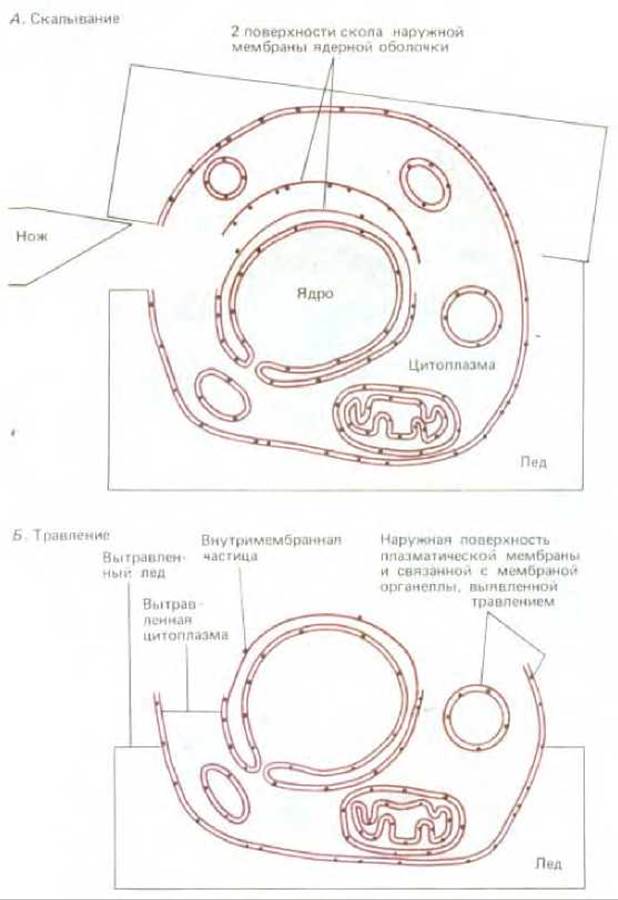
Рис. 4-23. Электронная микроскопия по методу замораживания-травления. Замороженный образец раскалывают ножом (А). Затем, сублимируя воду под вакуумом, уменьшают слой льда и обнажают таим образом поверхность клетки (Б). После этого готовят реплику все еще замороженной поверхности (как описано в подписях к рис. 4-21) и проводят исследование с помощью просвечивающего электронного микроскопа.
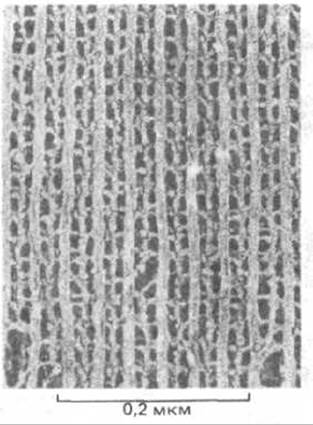
Рис. 4-24. Регулярно уложенные белковые филаменты в мышце насекомых. Для получения этого изображения мышечные клетки были заморожены в жидком гелии, расколоты через цитоплазму и подвергнуты глубокому высушиванию. Затем была приготовлена металлическая реплика, которую изучали при большом увеличении. (С любезного разрешения Roger Cooke, John Heuser).
Другой важный метод электронной микроскопии - метод «замораживания-травления» - используется для изучения внешней поверхности клеток и мембран. В данном случае клетки замораживают при очень низкой температуре и замороженный блок раскалывают лезвием ножа. Содержание льда вокруг клеток (и в меньшей степени внутри клеток) понижают возгонкой воды в вакууме при повышении температуры (процесс называют вакуумной сушкой) (рис. 4-23). Участки клетки, подвергнутые такому травлению, затем оттеняют (как было показано ранее) для приготовления платиновой реплики.
Метод замораживания - травления не позволяет использовать криппротекторы, поскольку они не летучи и по мере возгонки воды остаются в образце. Чтобы добиться высокого качества изображения, необходимо препятствовать образованию больших кристаллов льда. Это возможно при ускоренном замораживании образца (при скорости замораживания выше 20° С в миллисекунду). Один из методов такого быстрого замораживания состоит в использовании специального устройства. быстро сближающего образец с медным блоком, охлажденным до — 269 °С жидким гелием. Особенно впечатляющие результаты получают после глубокого травления быстро замороженных клеток. Этот метод позволяет выявлять структуры внутреннего содержимого клеток, демонстрируя их трехмерную организацию с исключительной четкостью (рис. 4-24).
Поскольку в этом случае в микроскопе под вакуумом наблюдают не образцы, а реплики, полученные после оттенения металлом, методы замораживание - скалывание и замораживание - травление можно использовать для изучения замороженных нефиксированных клеток и исключить риск проявления артефактов, вызванных фиксацией.
4.1.12. Методы негативного контрастирования и криоэлектронной микроскопии обеспечивают высокое разрешение при анализе макромолекул
Используя для контрастирования оттенение солями тяжелых металлов, можно наблюдать в электронный микроскоп изолированные макромолекулы, например, ДНК или большие белки (см. рис. 4-20), а после негативного контрастирования разрешению поддаются даже мельчайшие детали. При приготовлении образцов для негативного контрастирования исследуемые молекулы наносят на тонкую пленку углерода (практически прозрачную для электронов), затем ее смачивают концентрированным раствором солей тяжелых металлов, например, уранилацетата. После высушивания образца тонкая пленка солей тяжелых металлов равномерно покрывает углеродную подложку, за исключением участков, занятых адсорбированными макромолекулами. Вещество макромолекул более проницаемо для электронов по сравнению с прилежащими участками, покрытыми солями тяжелых металлов; за счет этого возникает обращенное или негативное изображение молекулы. Негативное окрашивание используется особенно эффективно для наблюдения больших агрегатов макромолекул (вирусы, рибосомы) либо для наблюдения субъединичной структуры белковых филаментов (рис. 4-25).
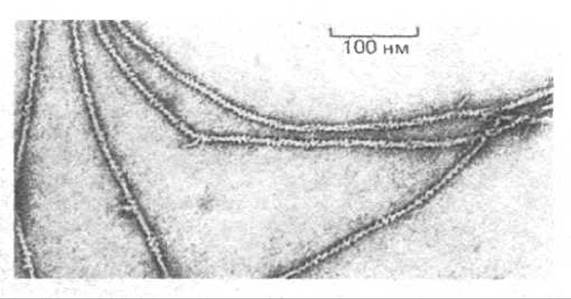
Рис. 4-25. Электронные микрофотографии негативно окрашенных актиновых нитей. Диаметр каждой из этих нитей составляет примерно 8 нм. При тщательном исследовании видно, что актиновые нити состоят из двух спирально закрученных цепей глобулярных молекул актина. (С любезного разрешения Roger Craig.)
Методы негативного контрастирования и оттенения обеспечивают высококонтрастное изображение поверхности небольших скоплений макромолекул, но разрешение этих методов ограничено размерами металлических частиц, образующих тень, либо молекул красителей, состоящих из солей тяжелых металлов, которые лишь грубо очерчивают поверхность молекулы или макромолекулярного ансамбля. Однако в настоящее время можно наблюдать с высоким разрешением даже внутренние детали трехмерных структур, таких, как вирусы. Для этого используют метод криоэлектронной микроскопии, где очень тонкий (примерно, 100 нм) быстро замороженный слой влажного образца помещают на микроскопическую решетку. С помощью специального приспособления гидратированный образец удерживают при — 160°С в вакууме микроскопа. Таким способом можно наблюдать материал практически непосредственно: без фиксации, окраски и сушки. Гомогенность витрифицированного водного слоя и использование недофокусированного фазового контраста позволяют получать удивительно четкие изображения таких неокрашенных образцов (рис. 4-26).
Вне зависимости от использованных методов отдельные белковые молекулы дают в электронном микроскопе слабые и плохо различимые изображения. Попытки извлечь информацию за счет удлинения времени наблюдения или повышения интенсивности освещающего луча тщетны, поскольку при этом имеет место разрушение наблюдаемого объекта. Для анализа деталей молекулярной структуры необходимо объединять информацию о многих молекулах, чтобы избежать случайных ошибок, исходящих от индивидуальных изображений. Такой подход приемлем для изучения вирусов или белковых филаментов, отдельные субчастицы которых представлены в виде регулярно повторяющихся элементов; он пригоден также для изучения любых веществ, которые могут быть расположены в двумерной кристаллической решетке, где значительное число молекул сохраняют одинаковую ориентацию или разделены одинаковыми промежутками. Используя электронные микрофотографии структур, ориентированных подобным образом, можно применить методы обработки изображения и высчитать среднее изображение отдельных молекул, выявив детали, приглушенные случайным «шумом» исходного снимка.

Рис. 4-26. Вирус леса Семлики в тонком слое неокрашенной вирифицированной воды (криоэлектронная микроскопия при — 160 °С.) С помощью электронной обработки микрофотографий можно получить трехмерное изображение с высоким разрешением. (С любезного разрешения Jacques Duboshet; см. также S.D. Fuller, Cell, 48, 923-934, 1987.)
Реконструкции изображения по этому методу позволяют получить детальную структуру оболочки вируса с разрешением в 3,5 нм, а детали формы индивидуальных макромолекул можно исследовать с разрешением 0,5 нм (5 А). Но даже наиболее изощренные методы электронной микроскопии не дают возможность полностью описать молекулярную структуру, поскольку атомы в молекулах разделены расстоянием около 0,10,2 нм. Для изучения молекулярной структуры макромолекул на атомарном уровне необходимы иные методы - методы. в которых вместо электронов используются рентгеновские лучи.
4.1.13. Детальная структура молекул, образующих кристаллическую решетку, может быть рассчитана на основании полученных дифракционных картин [12]
Рентгеновские лучи, подобно свету, являются одной из форм электромагнитного излучения, но вследствие того, что длина волны рентгеновских лучей значительно короче, их применение позволяет разрешить значительно более мелкие детали. Однако в отличие от видимого света или потока электронов, рентгеновские лучи нельзя сфокусировать и после их прохождения через образец получить обычное изображение. Однако структуру образца можно выявить, используя метод дифракции рентгеновских лучей.
Сперва рассмотрим одиночный объект (например, отдельную молекулу), расположенный на пути любого излучения, длина волны которого меньше размеров объекта. Объект будет рассеивать часть излучения. Рассеянное излучение можно рассматривать как набор перекрывающихся волн, каждая из которых отражается разными участками объекта. Если волны перекрываются, они подвергаются интерференции и возникает распределение излучения, известное как дифракционная картина. Дифракционная картина может быть зарегистрирована на фотопластинке, помещенной на некотором расстоянии от предмета, или представлена с помощью количества рассеянного излучения, отраженного объектом в разных направлениях (рис. 4-27). Форма дифракционной картины определяется структурой объекта. С другой стороны, исходя из полного описания дифракционной картины, можно теоретически рассчитать структуру данного объекта. Опыт показывает, что дифракционная картина для отдельной молекулы чересчур слаба и недостоверна, и потому ее нельзя использовать в качестве отправной точки для теоретического анализа.
Предположим, что множество идентичных объектов расположено в кристаллической решетке и на них направлен пучок каких-то лучей (рис. 4-28). В этом случае общее количество рассеянного излучения значительно выше. Однако излучение, рассеянное одной молекулой, будет интерферировать с излучением, рассеянным другими молекулами. В некоторых направлениях индивидуальные рассеянные лучи будут усиливаться, образуя на дифракционной картине яркое пятно.

Рис. 4-27. Рассеивание излучения одиночным объектом, размеры которою соизмеримы с длиной волны излучения. Излучение, падающее на объект, рассеивается в разных направлениях и с разной интенсивностью. Интенсивность излучения, рассеиваемого в данном направлении, зависит от интерференции излучения, рассеиваемого различными участками объекта. О результирующей интенсивности рассеивания во всех возможных направлениях можно судить по числу окрашенных лучей на диаграмме.
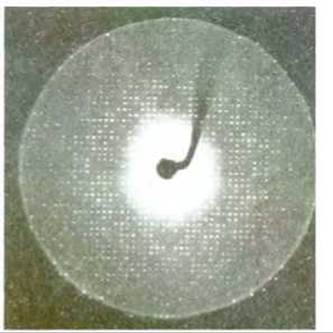
Рис. 4-29. Часть рентгенограммы кристалла белка. Именно этот кристалл был использован для определения расположения атомов в молекуле протеолитического фермента трипсина. (С любезного разрешения Robert Stroud.)
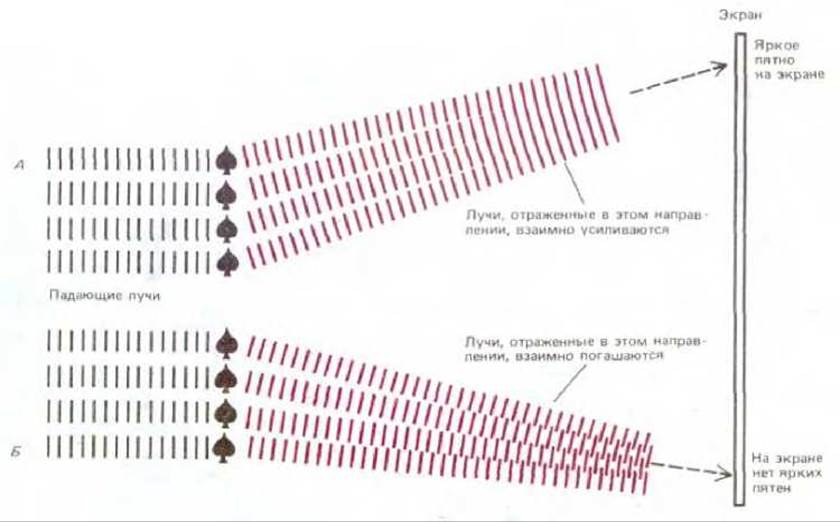
Рис. 4-28. Рассеивание излучения кристаллом. Если многие идентичные объекты расположены в виде кристаллической решетки, излучение, рассеиваемое каждым объектом, интерферирует с излучением, рассеиваемым другими объектами. Отдельные отраженные лучи рассеиваются только в определенных направлениях (в зависимости от пространственного расположения объекта в решетке), образуя яркие пятна. Интенсивность данного яркого пятна зависит от интенсивности, с которой каждый объект в решетке мог бы рассеивать излучение в данном направлении, если бы его исследовали в отдельности, как это показано на рис. 4-27.

Рис. 4-30. Кристаллы фермента гликоген-фосфорилазы при наблюдении в световой микроскоп. (С любезного разрешения Robert Fletterick.) дифракционная картина кристаллической решетки будет состоять из множества ярких пятен различной интенсивности (рис. 4-29). Относительная интенсивность различных пятен в дифракционной картине зависит от способности различных объектов в решетке рассеивать излучение. В действительности интенсивность данного пятна пропорциональна интенсивности излучения, которое будет отражаться в данном направлении от характерного одиночного объекта. Таким образом, положение пятен в дифракционной картине зависит от расположения объекта в системе, а их интенсивность дает информацию о внутренней структуре типичного объекта. Более того, такая информация является точной и достаточной, поскольку она была получена путем объединения вкладов множества равноценных источников. На самом деле, пользуясь довольно полным описанием дифракционной картины такой решетки, можно зачастую вычислить структуру отдельных объектов, образующих кристаллическую решетку.
4.1.14. Дифракция рентгеновских лучей дает возможность выявить трехмерную организацию атомов в молекулах [13]
Если для анализа молекулярной структуры предполагается использовать дифракционные картины, то излучение, претерпевающее дифракцию, должно иметь длину волны более короткую, чем расстояние между атомами в молекуле. Длина волны рентгеновских лучей около 0,1 нм (что соответствует диаметру атома водорода), и поэтому данный тип излучения идеально подходит для анализа расположения индивидуальных атомов в молекулах. Такую задачу нельзя решить даже на самых современных электронных микроскопах. Существенным преимуществом рентгеновских лучей является высокая (выше, чем у электронов) проникающая способность. Это делает пригодными для анализа более толстые образцы. И наконец, поскольку в данном случае использование вакуума не предусмотрено, можно изучать толстые водосодержащие образцы. Вследствие этого исключаются артефакты, возникающие в процессе приготовления образца.
Для достижения высокого разрешения необходимо иметь кристаллы с высокой степенью упорядоченности (рис. 4-30). По мере прохождения через образец рентгеновские лучи рассеиваются электронами атомов, составляющими образец. Поэтому большие атомы с большим количеством электронов рассеивают рентгеновские лучи более эффективно, чем небольшие атомы, так что атомы С, N, О, Р регистрируются гораздо более надежно, чем атомы Н; известно, что очень тяжелые атомы тоже рассеивают рентгеновские лучи очень эффективно. Процесс преобразования рентгенограммы в трехмерную структуру атомов, уложенных в молекулу, весьма сложен. Расшифровка рентгенограмм, образованных крупными и неупорядоченными молекулами белков, до 1960 года была невозможна (табл. 4-3). Эта процедура требует локализации и оценки интенсивности сотен тысяч пятен, равно как и фаз волн каждого пятна. В последние годы рентгеноструктурный анализ все более автоматизируется. Рассеянные рентгеновские лучи измеряются изощренными электронными детекторами, что существенно ускоряет процесс накопления данных, а мощные компьютеры выполняют множество необходимых вычислений. В настоящее время наиболее длительным этапом в подобном исследовании является этап получения подходящих кристаллов исследуемых макромолекул; зачастую на подбор оптимальных условий кристаллизации уходят годы. Но, несмотря на эти трудности, рентгеноструктурный анализ нашел широкое применение, поскольку до настоящего времени остается единственным методом определения детального расположения атомов в большинстве молекул. Имея хорошие кристаллы, можно рассчитать структуру белка с разрешением 0,3 нм и выявить не только основные закономерности расположения полипептидной цепи, но и некоторые более мелкие детали. Затратив значительные усилия, можно получить кристаллы высокого качества, что в свою очередь позволяет достичь разрешения 0,15 нм и определить расположение почти всех неводородных атомов в молекуле белка. Именно таким образом к настоящему времени были установлены структуры более сотни белков и нескольких малых молекул РНК и ДНК.
Таблица 4-3. Основные вехи в развитии метода рентгеноструктурного анализа и его применение в исследовании биологических молекул
|
1864 - Хоппе-Зейлер (Hoppe-Seyler) получил в кристаллическом виде гемоглобин и предложил для него название. 1895 - Рентген (Roentgen) наблюдал образование новой формы проникающей радиации при попадании катодных лучей (потока электронов) на металлическую мишень. Это излучение было названо Рентгеном Х-лучами (в русской научной литературе «рентгеновские лучи») 1912 - фон Лауэ (Von Laue) получил первую рентгенограмму, пропуская рентгеновские лучи через кристалл сульфида цинка 1912 - В. Л. Брэгг и В. Х. Брэгг (W. L. Bragg, W. Н. Bragg) обнаружили простую взаимосвязь между характером дифракционной картины и расположением атомов в кристалле 1926 - Самнер (Sumner) получил кристаллы уреазы из экстрактов канавалии мечевидной й показал, что эти белки обладают каталитической активностью 1931 - Полинг (Pauling) опубликовал свою работу «Природа химических связей», в которой уточнил правила ковалентного связывания 1934 - Бернал и Кроуфут (Bernall, Crowfoot) представили первую подробную рентгенограмму белка, полученную для кристаллов фермента пепсина 1935 - Паттерсон (Patterson) разработал аналитический метод определения расстояния между атомами по данным рентгеноструктурного анализа 1941 - Эстбюри (Astbury) получил первую рентгенограмму ДНК 1951 - Полинг и Кори (Pauling, Corey) обосновали существование двух основных типов укладки цепи L-аминокислот (в виде а-спирали и складчатого ß-слоя), которые были позже обнаружены во многих белках 1953 - Уотсон и Крик (Watson, Crick) предложили модель двойной спирали ДНК на основе рентгенограмм, полученных Франклин и Уилкинсом (Franklin, Wilkins) 1954 - Перутц (Perutz) и сотрудники разработали метод тяжелых атомов для решения проблемы фазы в кристаллографии белка 1960 - Кендрью (Kendrew) впервые подробно описал структуру белка (миоглобина кашалота) с разрешением 0,2 нм, а Перутц (Perutz) - структуру более крупного белка - гемоглобина, но в этом случае разрешение было несколько хуже 1966 - Филлипс (Phillips) впервые подробно описал структуру белка лизоцима 1976 - Ким, Рич, Клуг (Kim, Rich, Clugh) и сотрудники, использовав данные рентгеноструктурного анализа, подробно описали структуру тРНК 1977-78 - Холмс и Клуг (Holmes, Clugh) определили структуру вируса табачной мозаики (ВТМ), а Гаррисон и Россман (Harrison, Rossman) - структуру двух сферических вирусов |
Заключение
Для наблюдения клеток существует множество методов световой микроскопии. Окрашенные и фиксированные клетки можно наблюдать с помощью обычной оптики. Использование флуоресцентного микроскопа и меченых антител позволяет локализовать в клетках специфические молекулы. Клетки в естественном живом состоянии анализируют под фазово-контрастной, интерференционной или темнопольной оптикой. Светомикроскопические исследования живых клеток подкрепляются обработкой изображений с помощью электроники, что существенно усиливает чувствительности и увеличивает разрешение.
Определение подробной структуры мембран и органелл в клетках возможно только при высоком разрешении, которое дает просвечивающий электронный микроскоп. Просвечивающий электронный микроскоп также используют для определения формы индивидуальных макромолекул, оттененных тяжелыми металлами или негативным контрастированием. Однако точное расположение каждого атома в молекуле можно определить только после образования молекулами крупных кристаллов. В этом случае с помощью рентгеноструктурного анализа может быть рассчитана полная трехмерная структура молекулы.