Молекулярная биология клетки - Том 1 - Албертс Б., Брей Д., Льюис Дж., Рэфф М., Робертс К., Уотсон Дж. 1994
Введение в биологию клетки
Как изучают клетки?
Технология рекомбинантных ДНК
Основная проблема молекулярной биологии на сегодняшний день состоит в том, чтобы разобраться в тонких механизмах клеточных процессов. Мы обсудили несколько чувствительных методов очистки, анализа белков и слежения за ними в клетках. Этот последний раздел посвящен методам изучения структуры и функции клеточных ДНК. Классический подход подразумевает использование генетических методов, позволяющих судить о функции генов, анализируя фенотипы мутантных организмов и их потомства. Этот подход по-прежнему эффективен, но в последнее время он дополнен набором методов, которые в сумме известны как «технология рекомбинантных ДНК». Эти методы существенно расширили возможности генетических исследований, поскольку с их помощью удается проводить как прямой контроль, так и детальный химический анализ генетического материала. Используя методологию рекомбинантных ДНК, удается даже минорные клеточные белки получать в больших количествах и, следовательно, проводить тонкие биохимические исследования структуры и функции белка.
4.6.1. Технология рекомбинантных ДНК революционизировала клеточную биологию [37]
Еще не так давно, всего лишь в начале 70-х годов биохимики считали, что ДНК является наиболее сложным для исследования компонентом клетки. Чрезвычайно длинную, химически монотонную последовательность нуклеотидов в наследственном материале тогда можно было исследовать лишь с помощью косвенных методов - либо определяя структуру белка или РНК, либо с помощью генетического анализа. В настоящее время ситуация крайне изменилась. Если ранее анализ ДНК представлялся исследователям структуры биологических молекул крайне трудной задачей, то теперь, когда были разработаны новые методы анализа первичной структуры ДНК, такой анализ не составляет особого труда. В настоящее время можно вырезать отдельные участки ДНК, получать их практически в неограниченном количестве и определять последовательность нуклеотидов по нескольку сот нуклеотидов в день.
С помощью этих же методов можно по желанию экспериментатора изменить выделенный ген и ввести его вновь в геном культивируемых клеток или эмбрион животного (что несколько более сложно), где этот измененный ген начинает функционировать.
Технология рекомбинантных ДНК оказала существенное воздействие на всю клеточную биологию, позволяя исследователям решать задачи, которые раньше казались неразрешимыми, например определять функции многих вновь открытых белков и их индивидуальных доменов, расшифровывать сложные механизмы регуляции экспрессии генов у эукариот. С помощью методов генной инженерии удалось в большом количестве получить многие белки, участвующие в регуляции клеточной пролиферации и развитии. Применение этих методов должно принести успех в крупномасштабном промышленном производстве белковых гормонов и искусственных вакцин, на получение которых ранее затрачивали очень много сил и средств.
Технология рекомбинантных ДНК включает в себя набор методов - как новых, так и заимствованных из других дисциплин, например из генетики микроорганизмов (табл. 4-12). Наиболее важные среди них это:
1) специфическое расщепление ДНК рестрицирующими нуклеазами, что существенно ускоряет выделение и манипуляции с различными генами; 2) быстрое секвенирование всех нуклеотидов в очищенном фрагменте ДНК, что позволяет определить точные границы гена и аминокислотную последовательность, кодируемую им; 3) гибридизация нуклеиновых кислот, позволяющая выявлять специфические последовательности РНК или ДНК с большой точностью и чувствительностью на основании их способности связывать комплементарные последовательности нуклеиновых кислот; 4) клонирование ДНК: интересующий исследователя ДНК-фрагмент вводят в самореплицирующийся генетический элемент (плазмиду или вирус), который используют для трансформации бактерий. Бактериальная клетка после трансформации воспроизводит этот фрагмент во многих миллионах идентичных копий; 5) генетическая инженерия, посредством которой последовательности ДНК изменяют с целью создания модифицированных версий генов, которые затем вновь внедряют в клетки или организмы.
Таблица 4-12. Основные вехи в развитии технологии рекомбинантных ДНК
|
1869 - 1944 - |
Мишер (Miesher) впервые выделил ДНК Эвери (Avery) установил, что ДНК, а не белок, переносит генетическую информацию при трансформации бактерий |
|
1953- |
Уотсон и Крик (Watson, Crick) предложили модель двойной спирали ДНК, основанную на результатах рентгеноструктурного анализа, проведенного Франклин и Уилкинсом (Franklin, Wilkins) |
|
1961 - |
Мармур и Доти (Marmur, Doty) открыли явление ренатурации ДНК, установив точность и специфичность реакции гибридизации нуклеиновых кислот |
|
1962 - |
Арбер (Arber) впервые получил данные о существовании ферментов рестрикции ДНК, впоследствии выделенных и использованных для определения последовательности ДНК Натансом и Смитом (Nathans, Smith) |
|
1966 - |
Ниренберг, Очоа и Корана (Nirenberg, Ochoa, Khorana) расшифровали генетический код |
|
1967 - |
Геллерт (Gellert) открыл ДНК-лигазу - фермент, используемый для сшивания фрагментов ДНК |
|
1972-73 - |
В лабораториях Бойера, Коэна и Берга (Boyer, Cohen, Berg) и их коллег в Станфордском университете и в Калифорнийском университете в Сан-Франциско была разработана технология клонирования ДНК |
|
1975-77 - |
Сэнгер и Баррел (Sanger, Barrel), а также Максам и Гилберт (Махат, Gilbert) разработали методы быстрого определения нуклеотидной последовательности |
|
1981-82 - |
Пальмитер и Бринстер (Palmiter, Brinster) получили трансгенную мышь; Спрэдлинг и Рубин (Spradling, Rubin) получили трансгенные экземпляры дрозофилы |
Для того чтобы разобраться в технологии рекомбинантных ДНК, необходимо очень хорошо понимать природные механизмы, используемые клетками для репликации и расшифровки ДНК. Мы поэтому отложим детальное обсуждение клонирования генов и генетической инженерии. В гл. 5 эти вопросы будут разобраны после знакомства читателей с основными генетическими механизмами.
4.6.2. Рестрицирующие нуклеазы расщепляют ДНК в специфических участках нуклеотидных последовательностей [38]
Для защиты от молекул чужеродных ДНК, способных проникнуть в клетку и вызвать ее трансформацию, многие бактерии вырабатывают ферменты рестрицирующие нуклеазы, способные разрушить чужеродную ДНК. Каждый такой фермент опознает в ДНК специфическую последовательность из 4-6 нуклеотидов. Соответствующие последовательности в геноме самих бактерий замаскированы метилированием остатков А и С, но любая чужеродная молекула ДНК, попав в клетку, немедленно опознается нуклеазой, и обе цепи ее ДНК разрезаются (рис. 4-60). Из различных видов бактерий было выделено множество рестрицирующих нуклеаз. В настоящее время различными фирмами производится более 100 таких ферментов, большинство из которых опознает различные последовательности нуклеотидов.
Индивидуальная рестрицирующая нуклеаза способна разрезать двойную спираль ДНК любой длины с образованием серии фрагментов, называемых рестрикционными фрагментами (рестриктами). Сравнение размеров фрагментов ДНК, полученных после обработки определенного участка генома набором рестрицирующих нуклеаз, позволяет построить рестрикционную карту, на которой указано положение каждого сайта рестрикции относительно других рестрикционных участков (рис. 4-61) Поскольку рестрикционная карта отражает расположение определенной последовательности нуклеотидов в данном участке, сравнение таких карт для двух или более родственных генов позволяет приближенно оценить гомологию между ними. Отсюда следует, что можно проводить сравнение различных участков ДНК (сравнивая их рестрикционные карты) без определения их нуклеотидной последовательности. Например, сравнение рестрикционных карт, указанных на рис. 4-62, привело к выводу, что хромосомные участки, кодирующие цепи глобина у человека, орангутана и шимпанзе, сохранились практически в неизменном виде в течение последних 5-10 млн. лет, т.е. с тех пор, как эти виды дивергировали. Рестрикционные карты также важны для клонирования ДНК и генной инженерии; они позволяют локализовать ген на данном рестрикционном фрагменте. Объяснение этому вы найдете ниже.
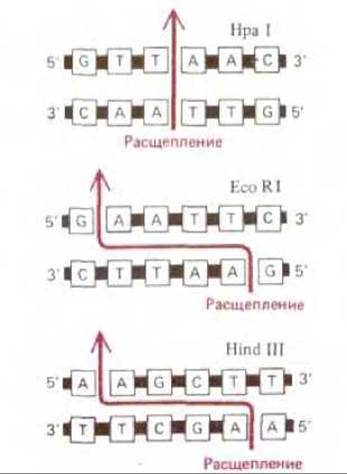
Рис. 4-60. Последовательность нуклеотидов в ДНК, узнаваемая тремя широко используемыми рестрицирующими нуклеазами (участки узнавания). Такие последовательности часто содержат шесть нуклеотидов и являются «палиндромными», т. е. последовательности нуклеотидов в них в обоих направлениях читаются одинаково. Две цепи ДНК разрезаются в участке узнавания или вблизи от него, причем во многих случаях разрез проходит через обе цепи не перпендикулярно, т. е. в одном месте, а наискосок, образуя в результате липкие концы, например Eco RI или Hind III. Рестрицирующие нуклеазы получают из различных бактерий: Hpa I - из Haemophilus parainfluenzae, Eco RI - из Escherichia coli, Hind III - из Haemophilus influenzae.

Рис. 4-61. Простой пример, иллюстрирующий взаимное расположение на двойной спирали ДНК участков узнавания для различных рестрицирующих нуклеаз (именуемых также «сайтами рестрикции»), совокупность которых образует рестрикционную карту. Заключение: фермент А расщепляет вблизи одного из концов молекулы. Фермент Б должен расщеплять вблизи того же конца либо вблизи другого конца. Размеры фрагментов, образующихся при расщеплении 2 ферментами исключают первое предположение и позволяют установить порядок сайтов рестрикции, указанный ниже
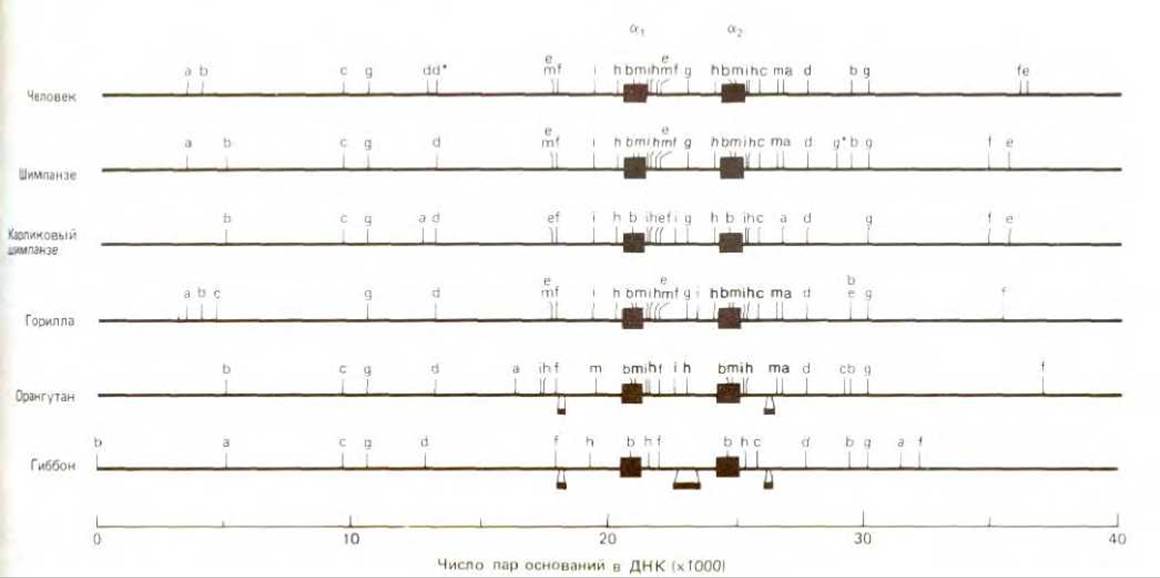
Рис. 4-62. Рестрикционная карта в кластере генов, кодирующих гемоглобин человека и различных приматов. Два окрашенных квадрата в каждой карте показывают положение участка, соответствующего гену глобина. Каждая буква указывает участок, расщепляемый различными рестрицирующими нуклеазами. Положение сайтов разреза определяли сравнением размеров фрагментов ДНК, образующихся при обработке ДНК различными рестрицирующими нуклеазами в отдельности и в различных сочетаниях. (С любезного разрешения Elisabeth Zimmer, Alan Wilson.)
4.6.3. Клонирование ДНК позволяет получать любые последовательности ДНК в большом количестве [39]
Многие рестрицирующие нуклеазы вносят разрывы в две цепи ДНК со смещением на несколько нуклеотидов, так что на концах фрагментов образуются короткие одноцепочечные участки. Эти одноцепочечные концевые участки обладают способностью образовывать комплементарные пары оснований с любым другим одноцепочечным участком, полученным с помощью того же фермента, и потому их называют липкими концами (рис. 4-63). Липкие концы, образованные рестрикционными ферментами, позволяют легко соединить два любых фрагмента ДНК воедино при условии, что эти фрагменты образовались после действия одной и той же рестрицирующей нуклеазы (рестриктазы) (либо иной нуклеазы, которая создает такие же липкие концы). Таким образом, фрагмент ДНК любого происхождения можно встроить в очищенную ДНК автореплицирующегося генетического элемента, которым, как правило, является плазмида или бактериальный вирус. Бактериальный клон, содержащий такую плазмиду или вирус, можно сравнить с фабрикой по производству этого фрагмента ДНК. Исходный фрагмент может происходить прямо из геномной ДНК, или из кДНК (комплементарной ДНК), т. е. из ДНК, полученной копированием матричной РНК. Эти методы детально обсуждаются в гл. 5.
Первый этап получения клонов геномной ДНК обычно включает выделение ДНК и ее расщепление рестриктазой. При этом возникает громадное количество различных фрагментов ДНК, например, в случае генома млекопитающих образуется от 105 до 107 фрагментов. В процессе клонирования исследователь получает миллионы клеточных колоний (клонов), большая часть которых содержит различные фрагменты ДНК. Наиболее сложным этапом клонирования является выявление именно того клона, который содержит нужный фрагмент ДНК.
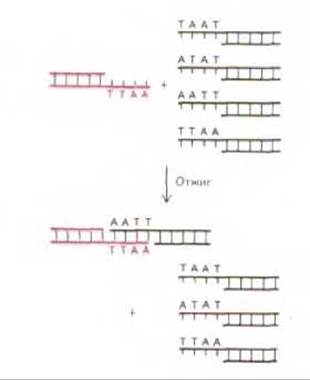
Рис. 4-63. Многие виды рестрицирующих нуклеаз позволяют получать фрагменты ДНК с липкими (одноцепочечными) концами. Фрагменты ДНК, обладающие такими концами, соединяются с помощью комплементарного взаимодействия пар оснований в области липких концов, как показано на схеме. Два фрагмента ДНК, объединяющиеся здесь, были получены с помощью рестрицирующей нуклеазы Eco.Rl (см. рис. 4-60).
Получение клонов кДНК начинается с выделения мРНК из клеток, Эта мРНК затем используется в качестве матрицы для обратной транскриптазы, фермента, синтезируемого определенными вирусами, ДНК которых возникает в процессе копирования последовательности РНК (процесс обратный по сравнению с обычным процессом копирования, в котором РНК синтезируется с ДНК-матрицы). Фермент создает ДНК-копию (кДНК) каждой из представленных молекул РНК. Эти одноцепочечные молекулы ДНК затем превращаются в двухцепочечные (см. разд. 5.6.3). Их клонируют с помощью методов, подобных тем, что были использованы для клонирования фрагментов геномной ДНК.
Следует иметь в виду, что существует принципиальное различие между геномными и кДНК-клонами, которое объясняется, в частности, характерным для высших эукариот процессом сплайсинга (см. разд. 9.4.8). По всей вероятности, именно клоны кДНК содержат непрерывные участки нуклеотидной последовательности, кодирующей белки.
4.6.4. Метод гель-электрофореза позволяет быстро фракционировать молекулы ДНК разного размера [40]
В начале 70-х годов было показано, что с помощью методов гель-электрофореза, которые оказались столь полезными для анализа белковых цепей, можно точно определить длину и чистоту молекул ДНК. Этот метод гораздо проще, чем модификация, используемая для белков; каждый нуклеотид в молекуле нуклеиновой кислоты уже обладает отрицательным зарядом, и поэтому нет необходимости добавлять отрицательно заряженный детергент ДСН, который «заставляет» белковые молекулы двигаться к положительному электроду. Были разработаны специальные полиакриламидные гели, с помощью которых удается разделить фрагменты ДНК длиной до 500 нуклеотидов, отличающиеся даже на один нуклеотид (рис. 4-64, А). К сожалению, поры в полиакриламидном геле для больших ДНК слишком малы, для их разделения по размеру были разработаны специальные гели на основе агарозы (полисахарид, выделяемый из морских водорослей) (рис. 4-64, Б). Оба этих метода разделения ДНК широко используются для аналитических и препаративных целей.

Рис. 4-64. Гель-электрофорез является мощным методом разделения молекул ДНК по их размерам. В трех указанных примерах электрофорез проводили сверху вниз, так что более крупные молекулы ДНК находятся в верхней части геля. А. Мелкопористый полиакриламидный гель был использован для фракционирования отдельных цепей ДНК. В диапазоне размеров от 10 до 500 нуклеотидов можно делить молекулы ДНК, отличающиеся лишь одним нуклеотидом. В этом случае на дорожки 1-4 нанесены продукты четырех независимых реакций секвенирования ДНК, где на концах цепи присутствуют дидезоксирибонуклеотиды G, А, Т и С (в скобках см. рис. 4-68); так как в этих реакциях используют молекулы ДНК, предварительно меченные радиоизотопами, расположение этих радиоизотопов можно выявить радиоавтографически, как указано на рисунке. Б. Среднепористый агарозный гель используется для разделения двухцепочечных молекул ДНК. Этот метод наиболее удобен для разделения молекул длиной от 300 до 5000 пар нуклеотидов. Именно такую длину имеет рестрикционные фрагменты бактериофага, выявляемые по флуоресценции после окраски бромистым этидием. В. Метод пульс-электрофореза в агарозном геле использован для разделения 16 различных хромосом дрожжей (Saccharomyces cerevisiae), размеры которых варьируют от 220000 до 2500000 пар нуклеотидов. В этих гелях можно разделять молекулы длиной до 107 пар нуклеотидов. (А - с разрешения Linder Laufer, Peter Walter; Б - с разрешения Ken Kreutzer, В - с разрешения D. Wolrath, A.W. Davies - Nucl. Acid Res., 15, 7876, 1988.)
Недавно была предложена модификация гель-электрофореза в агарозном геле, названная электрофорез в пульсирующем электрическом поле или пульс-электрофорез. С ее помощью удается разделять очень большие, можно сказать громадные молекулы ДНК. Обычный гель- электрофорез не позволяет разделить такие молекулы ввиду постоянства электрического поля, которое придает молекулам змеевидную конфигурацию. Обладающие такой конфигурацией молекулы движутся в гелях с постоянной скоростью вне зависимости от длины молекул. Если же направление электрического поля будет часто меняться, скорость движения молекул будет определяться их способностью переориентироваться согласно этому изменению. Такой процесс у больших молекул занимает значительно больше времени, вследствие чего они будут отставать. На гелях после пульс-электрофореза целые хромосомы бактерий или дрожжей выявляются в виде отдельных полос (рис. 4-64, В), и поэтому можно легко определить хромосомные перестройки. Более того, используя гибридизацию молекул клонированной ДНК данного геля для поиска комплементарных последовательностей в геле, удалось картировать множество генов у дрожжей (см. разд. 4.6.8).
Если не произвести мечение или окраску ДНК, полосы в агарозных или полиакриламидных гелях останутся невидимыми. Один из самых эффективных методов окраски ДНК состоит в выдерживании геля после электрофореза в растворе красителя бромистого этидия, который флуоресцирует под ультрафиолетовым светом после связывания с ДНК (рис. 4-64, Б и В). Еще более чувствительный метод детекции основан на включении радиоизотопов в молекулы ДНК до электрофореза; для этого обычно используют 32Р, который включается в фосфаты ДНК и испускает ß-частицу достаточно высокой энергии, чтобы её можно было выявить с помощью метода радиоавтографии (рис. 4-64, A).
4.6.5. Очищенные молекулы ДНК можно метить радиоизотопами in vitro [41]
Для мечения очищенных молекул ДНК радиоизотопами широко используются два метода. В первом случае в молекулу ДНК с помощью ДНК-полимеразы I E.coli вводят радиоактивно меченные нуклеотиды (рис. 4-65, А). При этом получают радиоактивные «ДНК-зонды», используемые в реакциях гибридизации нуклеиновых кислот (см. ниже). Во втором методе фермент полинуклеотидкиназа из бактериофага используется для переноса отдельных фосфатов, меченных 32Р, с АТР на 5'-конец каждой из цепей ДНК (рис. 4-65, Б). Поскольку каждая из цепей ДНК метится с помощью киназы только одним атомом 32 Р, молекулы ДНК обычно недостаточно радиоактивны, чтобы использоваться в качестве зондов ДНК; однако то, что помеченными цепи ДНК оказываются только по одному концу, делает их очень удобными для секвенирования и «футпринтирования», что будет обсуждаться ниже.
4.6.6. Выделенные фрагменты ДНК можно легко секвенировать [42]
В конце 70-х годов были разработаны методы для простого и быстрого определения последовательности нуклеотидов (секвенирования) любых очищенных фрагментов ДНК. Вслед за этим были определены полные последовательности нуклеотидов многих генов млекопитающих, включая гены, кодирующие гемоглобин, инсулин и цитохром с. Объем информации о последовательностях ДНК столь велик (многие миллионы нуклеотидов), что для хранения и анализа имеющихся данных необходимо использовать компьютеры. Секвенировано несколько протяженных последовательностей ДНК, содержащих более 105 пар нуклеотидов; среди них полный геном вируса Эпштейна-Барр (вызывающего у человека инфекционный мононуклеоз), а также полный геном хлоропластов растений. В настоящее время широко используются два различных метода секвенирования ДНК; принципы, лежащие в основе химического метода иллюстрированы рис. 4-66 и 4-67, ферментативный метод объясняется на рис. 4-68.
Эти методы настолько быстры и надежны, что, когда перед исследователем стоит задача выяснения последовательности аминокислот в белке, оказывается целесообразным провести секвенирование соответствующего гена и реконструировать последовательность аминокислот на основании генетического кода. Хотя считывание любой ДНК может происходить в принципе с шестью различными рамками считывания (по три в каждой цепи), истинную рамку считывания определяют по следующему свойству: обычно это единственная рамка считывания, в которой стопкодоны встречаются редко (см. разд. 5.1.6). Чтобы убедиться в том, что выводя последовательность аминокислот в белке из последовательности нуклеотидов в соответствующем гене, мы не ошиблись.

Рис. 4-65. Два ферментативных метода, используемые обычно для получения радиоактивных молекул ДНК. А, ДНК-полимеразой I метят все нуклеотиды в молекуле ДНК, что позволяет получить высокорадиоактивные ДНК-зонды. Б. Полинуклеотид-киназа метит только 5'-концы ДНК. Если мечение ДНК сочетают с обработкой рестрикционной нуклеазой, как указано на рисунке, фракция молекул ДНК, содержащих меченные по 5'-концу отдельные цепи, может быть довольно легко зарегистрирована.
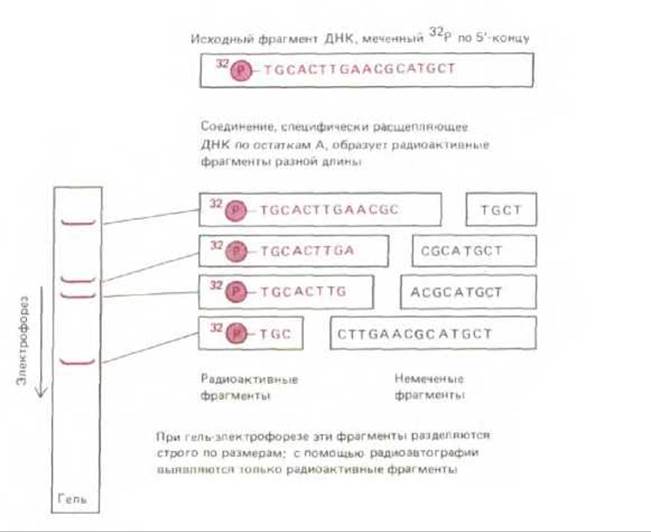
Рис. 4-66. Семейство меченных по 5'-концу фрагментов ДНК, полученных в результате рестрикции по определенному нуклеотиду (в данном случае основание А). Анализируемая цепь ДНК - продукт денатурации двухцепочечной молекулы, выделенной с помощью метода, описанного на рис. 465, Б. Эту цепь подвергли мягкой химической обработке, удаляющей из цепи некоторое количество одного из четырех нуклеотидов; при этом большинство таких нуклеотидов остаются в цепи. Поскольку только фрагменты, указанные на рисунке слева, содержат 5'-конец и 32Р-фосфатную группу на ней, именно эти фрагменты регистрируются после радиоавтографии геля. Данная процедура лежит в основе химического метода секвенирования ДНК, описанного на рис. 4-67. следует прямо секвенировать небольшой участок аминокислотной последовательности в очищенном белке.
Модификация метода секвенирования ДНК, представленная на рис. 4-66 и 4-67, может быть использована для выявления в ДНК последовательностей, опознаваемых ДНК-связывающими белками. Связывание этих белков с регуляторными участками ДНК (которые обычно локализованы вне кодирующих участков генов), по-видимому, играет важную роль в определении того - какие именно гены активны в данном типе клеток. Понимание функции этих белков чрезвычайно важно для идентификации специфических последовательностей, с которыми они связываются. Для выявления таких последовательностей обычно используют метод, именуемый ДНК-футпринтинг. Сперва очищенный фрагмент ДНК метят по одному концу 32Р и затем расщепляют с помощью нуклеазы или химического соединения, делающего случайные разрезы в двойной спирали ДНК. Фрагменты, которые образуются из меченой цепи, разделяют на геле и выявляют на радиоавтографе; после этого сравнивают расположение полос ДНК, образуемых в присутствии и в отсутствие ДНК-связывающих белков. Если связывание произошло, нуклеотиды в сайте расщепления оказываются защищенными от действия нуклеазы. В результате меченые фрагменты, содержащие участок нуклеазы. В результате меченые фрагменты, содержащие участок связывания, отсутствуют, и на геле возникает промежуток, не содержащий фрагментов ДНК, именуемый «футпринт», или «отпечаток ноги» (рис. 4-69, А). На рис. 4-69, Б представлен футпринт белков, активируют их транскрипцию эукариот.

Рис. 4-67. Химический метод секвенирования ДНК. Процедура, описанная на рис. 4-66, выполняется одновременно для четырех одинаковых проб ДНК. При этом используют химические агенты, расщепляющие ДНК в первом случае по Т, во втором по С, в третьем по G и четвертом по А. Полученные образцы подвергают электрофорезу на параллельных дорожках одного геля, как это указано на рис. 4-64, А. Анализируя результаты электрофореза, можно определить последовательность ДНК. Так, первая снизу полоса соответствует нуклеотиду, расположенному на 5'-конце. При этом определяют, на какой из дорожек расположена полоса, - в данном случае это Т. Для определения полной последовательности эту же процедуру выполняют на уровне второй, затем третьей полосы и так далее. В данном случае метод идеализирован; на самом деле химическая обработка менее специфична, чем указано здесь.
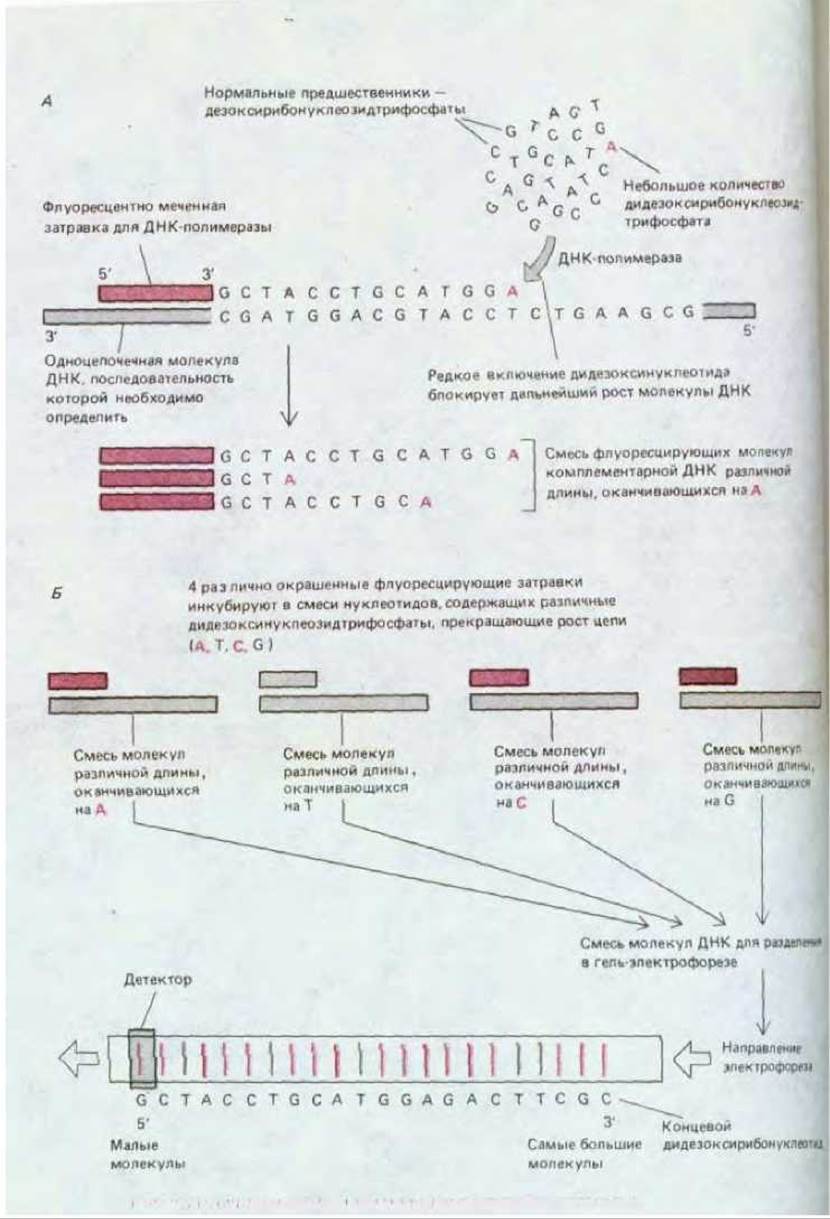
Молекулы, оканчивающиеся каждым из дидезоксирибонуклеотидов, выявляют в виде окрашенных полос по соответствующим окрашенным затравкам. Таким образом можно прямо прочесть последовательность цепи комплементарной ДНК по мере прохождения детектором полос А
Рис. 4-68. Метод секвенирования нуклеиновых кислот, основанный на энзиматическом введении нуклеотида, терминирующего цепь. Ключевым для этого метода является использование дидезоксирибонуклеозидтрифосфатов, в которых дезоксирибоза-3'-ОН, представленная в нормальных нуклеотидах, в данном случае отсутствует; внедряясь в цепь ДНК, такой модифицированный нуклеозид блокирует присоединение следующего нуклеозида. А. Синтез in vitro молекулы ДНК в присутствии затравки, а также небольшого количества одного из таких модифицированных нуклеозидов приводит к образованию «лесенки» фрагментов ДНК, представленных на рис. 4-66. Если для получения таких фрагментов применяют радиоактивную ДНК, проводят четыре различные реакции синтеза, каждая из которых использует различные нуклеозиды, терминирующие цепь, а электрофоретический анализ проводят на четырех параллельных дорожках геля, можно определить последовательность нуклеотидов в ДНК (см. рис. 4-67, а также рис. 4-64, А). Б. Более современная модификация метода, в которой четыре набора фрагментов, меченные различным образом, автоматически анализируются по флуоресценции в процессе движения по одной дорожке геля.

Рис. 4-69. Метод футпринтирования ДНК. А. Белок плотно связывается со специфическим участком ДНК из восьми нуклеотидов и защищает его от расщепляющего агента. Если реакция выполняется без белка, связывающегося с ДНК, на геле проявится полный набор полос (не указано). Б. Реальный футпринт, использованный для определения участка связывания для белка человека, стимулирующего транскрипцию некоторых генов эукариот. Из результатов следует, что данный сайт расположен за 60 нуклеотидов до сайта инициации синтеза РНК. В качестве расщепляющего агента использовали низкомолекулярное железосодержащее органическое вещество. Это вещество в норме расщепляет каждую из фосфодиэфирных связей практически с равной частотой. (Б - с разрешения Michelle Savadogo, Robert Roeder.)
4.6.7. Реакция гибридизации нуклеиновых кислот - чувствительный метод выявления специфических последовательностей нуклеотидов [43]
Если водный раствор ДНК нагреть до 100 °С и сильно защелочить (рН 13), то комплементарные пары оснований, удерживающие две цепи двойной спирали вместе, разрушатся и ДНК быстро диссоциирует на две цепи. Этот процесс, называемый денатурацией ДНК, ранее считала необратимым. Однако в 1961 году было обнаружено, что если комплементарные цепи ДНК выдержать при температуре 65 °С, они легко спариваются, восстанавливая структуру двойной спирали (процесс, получивший название ренатурации или гибридизации). Подобные процессы гибридизации могут происходить между двумя любыми одинарными цепями нуклеиновых кислот (ДНК—ДНК, РНК—РНК, ДНК—РНК; при условии, что они содержат комплементарные последовательности нуклеотидов.
Скорость формирования двойной спирали лимитируется вероятностью столкновения двух комплементарных последовательностей нуклеиновых кислот, что в свою очередь определяется их концентрацией в растворе. Скорость гибридизации может быть использована для определения концентрации любых последовательностей РНК или ДНК в смеси, содержащей и другие последовательности нуклеиновых кислот. Для этого теста необходимо иметь чистый одноцепочечный фрагмент ДНК, комплементарный к той последовательности, которую нужно обнаружить. Этот фрагмент ДНК можно получить клонированием, а если последовательность короткая, ее можно синтезировать химическими методами. В любом случае фрагмент ДНК интенсивно метят 32Р (см. рис. 4-65) с тем, чтобы можно было следить за включением этой молекулы в состав дуплексов в процессе реакции гибридизации, Одноцепочечная молекула ДНК, используемая здесь в качестве индикатора, называется ДНК- зонд; она может содержать от 15 до 1000 нуклеотидов.
Реакция гибридизации с использованием ДНК-зондов настолько чувствительна и избирательна, что с ее помощью можно идентифицировать последовательности, присутствующие в концентрации 1 молекула на клетку (рис. 4-70). Это позволяет определить, какое количество копий последовательности ДНК, комплементарной ДНК-зонду, присутствует в геноме клетки. Этот же метод весьма эффективен для поиска неидентичных, но родственных генов; например, после клонирования интересующих исследователя генов мыши или курицы, их последовательности могут быть использованы для поиска соответствующих генов в геноме человека.
ДНК-зонды применяют и в реакциях гибридизации с РНК до выявления экспрессии данного гена в клетках. В этом случае ДНК-зонд, содержащий часть последовательности гена, пытаются гибридизовать с РНК, выделенной из анализируемой клетки. Если гибридизация происходит, проводят количественное определение экспрессии. Более усовершенствованные методики предполагают обработку ДНК-зонда специфическими нуклеазами для обнаружения участков, гибридизующих с клеточной РНК. Таким образом можно определить начальные и концевые участки транскриптов РНК (рис. 4-71); этот же метод может быть полезен для выяснения точных границ участков, вырезаемых из транскриптов РНК в процессе сплайсинга РНК.
В процессе развития эмбриона происходит включение и выключение больших групп генов и этот процесс скоординирован. Гибридизация ДНК-зонда с клеточными РНК позволяет ответить на вопрос, работает или молчит определенный ген; более того, при изменении уровня экспрессии гена можно узнать, зависит ли это изменение от контроля,
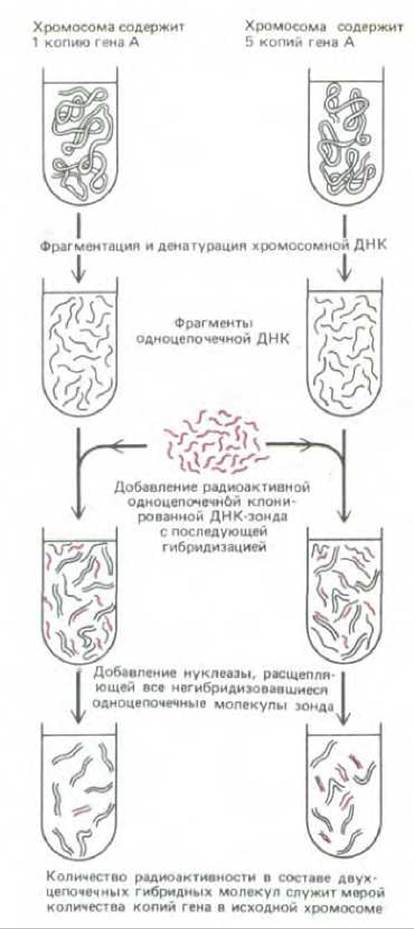
Рис. 4-70. Измерение количества копий определенного гена в образце ДНК с помощью гибридизации ДНК. Одноцепочечный радиоактивный фрагмент ДНК, используемый в таких экспериментах, называют ДНК-зондом. Хромосомная ДНК в данном случае не содержит радиоактивных атомов.
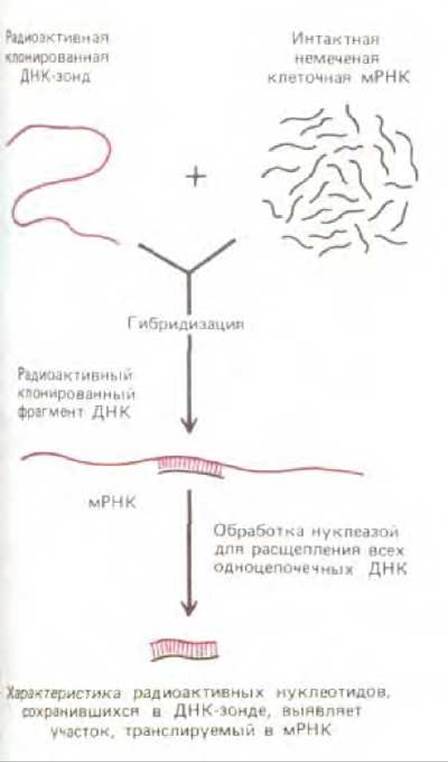
Рис. 4-71. Использование гибридизации нуклеиновых кислот для определения участка клонированного фрагмента ДНК, который транскрибируется в мРНК. Данный метод предполагает обработку нуклеазой, расщепляющей цепи ДНК, не спаренные с комплементарной цепью РНК. Этот метод позволяет точно выявлять начало и конец молекулы РНК. Аналогичные процедуры эффективны и для определения расположения нитронов (некодирующих последовательностей эукариотических генов).
действующего на уровне транскрипции ДНК, сплайсинга РНК или же трансляции зрелых молекул мРНК в белок. Методы гибридизации в современной клеточной биологии используются настолько широко, что даже трудно представить, как можно было бы без них изучать структуру генов и их экспрессию.
4.6.8. Методы Нозерн- и Саузерн-блоттинга позволяют гибридизовать молекулы нуклеиновых кислот, предварительно фракционированные с помощью электрофореза [44]
Для выявления молекул нуклеиновых кислот, последовательность которых комплементарна всему зонду или его участку, ДНК-зонды часто используются в сочетании с гель-электрофорезом. Множество различных молекул РНК и ДНК, содержащихся в смеси, фракционируют электрофорезом согласно размеру и после этого проводят реакцию гибридизации; если проба метит молекулы одного или нескольких размеров, можно быть уверенным, что гибридизация достаточно специфична. В некоторых случаях очень ценной является информация даже о размере гибридизуемых молекул ДНК. Это будет проиллюстрировано следующим примером.
Допустим, что перед исследователем стоит задача определить природу дефекта у мутантной мыши, синтезирующей аномально низкое количество альбумина (белка, который в норме секретируется в кровь клетками печени в значительных количествах). Для этого прежде всего необходимо взять образцы ткани печени у дефектных и нормальных мышей (последние служат в качестве контролей) и обработать клетки сильным детергентом для инактивации клеточных нуклеаз, которые в противном случае могут разрушить нуклеиновые кислоты. Затем отделяют РНК и ДНК от всех других компонентов клетки: присутствующие белки при этом полностью денатурируются, их удаляют последовательной экстракцией фенолом - мощным органическим растворителем. Нуклеиновые кислоты остаются в водной фазе. Чтобы их отделить от низкомолекулярных клеточных соединений, проводят осаждение спиртом. После этого ДНК отделяют от РНК, пользуясь их различной растворимостью в спиртах и обрабатывают высокоспецифическими ферментами (соответственно РНКазой или ДНКазой), чтобы освободиться от нежелательных примесей нуклеиновых кислот.
Для анализа РНК, кодирующих альбумин, с помощью ДНК-зонда используется метод Нозерн-блоттинга. На первом этапе с помощью гель-электрофореза, фракционируют интактные молекулы РНК дефектных и контрольных клеток печени и получают набор полос. Для того чтобы молекулы РНК, содержащиеся в геле, сделать более доступными ДНК-зонду, осуществляют перенос (блоттинг) фракционированных молекул РНК из геля на лист нитроцеллюлозы. На следующем этапе лист нитроцеллюлозы инкубируют с раствором, содержащим меченый ДНК-зонд. Полосы РНК, гибридизующиеся с зондом, выявляют методом радиоавтографии (рис. 4-72). Известно, что скорость движения молекул нуклеиновых кислот в геле зависит от их размера: при электрофорезе малые молекулы перемещаются быстрее, чем большие. Сравнивая скорость миграции молекул интересующего нас образца и молекул РНК известного размера (стандарты РНК), можно определить размеры каждой молекулы, связывающей зонд. При этом может оказаться, что клетки печени дефектных мышей синтезируют РНК альбумина в нормальных количествах и размер этих молекул соответствует норме. Если бы это было не так, выявлялось бы уменьшенное количество молекул РНК нормального альбумина. Возможен также вариант, когда молекулы РНК из дефектных клеток печени окажутся укороченными и вследствие этого будут перемещаться в геле быстрее, чем в норме. В этом последнем случае блот, содержащий дефектные молекулы РНК, можно подвергнуть повторной гибридизации с более чувствительными ДНК-зондами для выявления утраченных участков.
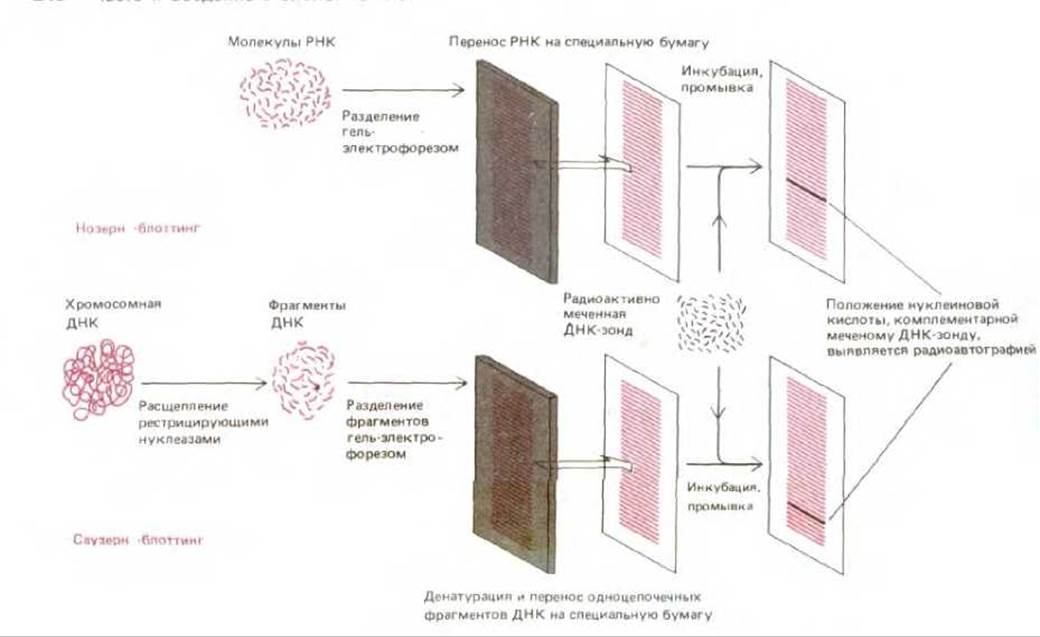
Рис. 4-72. Методы «Нозерн»- и «Саузерн»-блоттинга. После электрофоретического фракционирования смеси молекул ДНК или РНК в агарозном геле проводят перенос различных фрагментов нуклеиновых кислот на лист нитроцеллюлозы или найлона («блоттинг»). Этот лист затем инкубируют с радиоактивным ДНК-зондом в течение длительного времени в условиях, способствующих гибридизации. Затем лист тщательно промывают, так что радиоактивно меченными оказываются лишь те фрагменты, которые гибридизуются с ДНК-зондом. На радиоавтографе, полученном с листа нитроцеллюлозы, эти фрагменты выявляются в виде полос.
Для анализа структуры гена альбумина дефектных мышей был использован метод Саузерн-блоттинга. В данном случае вместо РНК анализируется ДНК. Изолированную ДНК сначала обрабатывают рестрицирующими нуклеазами, затем полученные фрагменты разделяют по размеру гель-электрофорезом и выявляют комплементарные ДНК-зонду альбумина с помощью переноса и гибридизации, как это описано для РНК (см. рис. 4-72). Повторяя эту процедуру с различными рестрицирующими нуклеазами, можно получить детальную рестрикционную карту генома в участке альбуминового гена (см. разд. 4.6.2). Анализируя эту карту, можно ответить на вопрос, несет ли альбуминовый ген у дефектных животных перестройки, например, делеции или инсерции коротких фрагментов ДНК.
4.6.9. Искусственные ДНК-зонды позволяют проводить дородовую диагностику наследственных болезней [45]
Пока микробиологи разрабатывали методы клонирования ДНК, химики-органики усовершенствовали методы синтеза коротких фрагментов ДНК. В настоящее время это делают с помощью приборов, способных автоматически синтезировать любые последовательности из 80 нуклеотидов в течение ночи. Умение получать молекулы ДНК заданной последовательности дает возможность перестраивать гены, что является важным аспектом генной инженерии (гл. 5).
Существенной областью применения ДНК-олигонуклеотидов является дородовая (пренатальная) диагностика наследственных заболеваний. Более 500 наследственных болезней человека связаны с нарушением какого-то одного гена. В большинстве случаев эти мутации рецессивны. Это означает, что болезнь развивается, если человек получает дефектные копии гена сразу от обоих родителей. Одна из задач современной медицины состоит в том, чтобы выявлять такие аномальные эмбрионы до рождения, информировать об этом мать и дать ей возможность прекратить беременность. Например, для серповидноклеточной анемии известна точная нуклеотидная замена в мутантном гене (последовательность GAG заменена на GTG в цепи ДНК, кодирующей ß-цепь гемоглобина). В данном случае синтезируют два олигонуклеотида. Один из них соответствует последовательности нормального гена в участке предполагаемых мутаций, другой несет замену, обусловливающую болезнь. В условиях когда эти последовательности достаточно коротки (примерно 20 нуклеотидов) и при температуре гибридизации, при которой стабильность сохраняют лишь точно совпадающие цепи, можно использовать радиоактивные зонды. Тест состоит в том, что из эмбриональных клеток, содержащихся в амниотической жидкости (ее получают в ходе процедуры, называемой амниоцентезом), выделяют ДНК и используют ее для Саузерн-блоттигна с радиоактивными ДНК-зондами. Дефектный эмбрион легко опознается, поскольку его ДНК будет гибридизоваться только с олигонуклеотидом, комплементарным мутантной последовательности ДНК. К сожалению, для большинства наследственных болезней дефект на уровне ДНК еще не расшифрован, однако круг заболеваний, для которых применяется дородовая диагностика, постоянно расширяется. Это стало возможно благодаря использованию феномена полиморфизма длины рестрикционных фрагментов. В данном случае с помощью гибридизации выявляют наличие или отсутствие определенных сайтов рестрикции, тесно сцепленных с дефектными генами.
4.6.10. Гибридизация позволяет выявлять и отдаленно родственные гены [46]
Возникновение новых генов в ходе эволюции связано с дивергенцией и дупликацией старых генов, а также объединением участков генов в новых комбинациях. По этой причине большинство генов имеют в геноме семейства родственные последовательности, часть которых, по- видимому, обладает и близкой функцией. Выделение ДНК-клона, соответствующего первому из членов такого генного семейства, - процедура весьма трудоемкая (разд. 5.6.5). Однако выделение остальных генов этого семейства упрощается, поскольку первый ген может быть использован в качестве зонда. Поскольку в родственных генах маловероятно присутствие идентичных последовательностей, гибридизацию с ДНК-зондами обычно выполняют в менее строгих условиях. Благодаря этому даже неполное соответствие последовательности зонда позволяет сформировать стабильную двойную спираль (рис. 4-73).
Хотя использование нестрогой гибридизации сопровождается повышением вероятности получения фальшивых сигналов от случайных участков гомологии коротких последовательностей в неродственных участках ДНК, такая гибридизация представляет собой одно из наиболее удачных применений технологии рекомбинантных ДНК. Например, этот подход привел к выделению всего семейства ДНК-связывающих белков, которые функционируют в качестве ключевых регуляторов экспрессии генов в ходе раннего эмбрионального развития Drosophila (см. разд. 16.5.19). Этот же подход был использован для идентификации родственных этому семейству генов из других организмов, включая человека.

Рис. 4-73. Сравнение нескольких модификаций метода гибридизации, отличающихся различной жесткостью условий. В реакции слева (жесткие условия) температура раствора поддерживается лишь на несколько градусов ниже температуры денатурации полностью комплементарной спирали ДНК (ее температуры плавления). В этих условиях спирали, совпадающие не полностью и формирующиеся при пониженной жесткости (см. справа), оказываются нестабильными. Справа - указаны условия гибридизации, используемые для поиска родственных генов, не полностью идентичных гену А.
4.6.11. Для локализации специфических последовательностей нуклеиновых кислот в хромосомах и клетках используют гибридизацию in situ [47]
Все макромолекулы клетки, и в том числе нуклеиновые кислоты, занимают в тканях и клетках строго определенное положение. Экстрагирование этих молекул из тканей или клеток путем гомогенизации приводит к потере той части информации, которая относится к расположению нуклеиновых кислот в клетках. Поэтому были разработаны методы для локализации специфических последовательностей нуклеиновых кислот in situ - в изолированных хромосомах, в определенных типах клеток, где ДНК- и РНК-зонды используются примерно так же, как меченые антитела. Этот метод называют гибридизацией in situ. Его применяют для анализа ДНК в хромосомах и РНК в клетках. Высокорадиоактивные зонды нуклеиновых кислот гибридизуют с хромосомами после кратковременного воздействия высоким рН с целью разделения пар оснований ДНК. Участки хромосом, связывающие радиоактивные зонды в процессе гибридизации, выявляются радиоавтографией. Пространственное разрешение этого метода может быт повышено при использовании не радиоактивно меченных, а химически меченных зондов ДНК. Как правило, при синтезе зондов используют нуклеотиды, содержащие боковую цепь биотина, и гибридизовавшиеся пробы выявляются при окраске стрептавидином (молекулы которого расположены в виде сети) или с помощью иных маркерных молекул (рис. 4-74). Были также разработаны методы гибридизации in situ, позволяющие судить о распределении специфических молекул РНК в клетках внутри тканей, В этом случае ткани не подвергают воздействию высоких значений рН, так что хромосомная ДНК остается в двухцепочечном состоянии и не может связывать зонд. Однако если ткань подвергнуть слабой фиксации,
РНК, содержащаяся в ней, при инкубации ткани с комплементарным ДНК-зондом обеспечивает возможность гибридизации. Таким способом удалось наблюдать, как реализуется дифференциальная активность генов Drosophila (рис. 4-75). Этот подход позволил существенно продвинуться в изучении молекулярных механизмов дифференцировки различных клеток эмбриона.
4.6.12. Методы рекомбинантных ДНК дают возможность изучать даже минорные белки клеток [48]
До недавнего времени изучение клеточных белков было ограничено лишь мажорными фракциями, т.е. белками, содержащимися в клетках в относительно большом числе. С помощью обычных методов хроматографии и электрофореза из нескольких сот граммов клеточной массы можно получить примерно 0,1 г (100 мг) одного из мажорных белков, составляющих 1% или более от всего количества клеточных белков. Этой массы белка вполне достаточно для изучения его аминокислотной последовательности, детального анализа биологической или ферментативной (если таковая имеется) активности и получения антител, которые могут быть использованы для локализации белков в клетках. Более того, когда удается вырастить подходящие кристаллы, то с помощью рентгеноструктурного анализа можно установить трехмерную структуру молекулы. Именно таким образом была определена структура и функция многих распространенных белков, в том числе гемоглобина, трипсина, иммуноглобулина и лизоцима.
Эукариотическая клетка содержит тысячи различных белков, но значительное большинство этих белков, и в том числе наиболее интересные, присутствуют в небольшом количестве. Некоторые из них иногда чрезвычайно трудно, если не невозможно, получить в чистом виде в количестве, превышающем несколько микрограмм. Разработка методов рекомбинантных ДНК сделала доступными любые клеточные белки (включая минорные белки) в больших количествах. Для этого клонируют ген нужного белка и затем встраивают его в специальную плазмиду, именуемую клонирующим вектором. Этот вектор сконструирован таким образом, что будучи введенным в бактерии, дрожжи или клетки млекопитающих соответствующего типа, он обеспечивает крупномасштабный синтез этого белка. Таким образом, если раньше для детальных структурных или функциональных исследований были доступны лишь немногие белки, в настоящее время практически все белки клетки могут быть предметом подобных исследований.

Рис. 4-74. Локализация гена на политенной хромосоме Drosophila с помощью гибридизации in situ с клонированным ДНК-зондом, меченным биотином. ДНК этой гигантской хромосомы частично денатурирована для гибридизации с ней зонда. После гибридизации и отмывки зонда, хромосомы обрабатывают ферментативным комплексом, в составе которого пероксидаза хрена конъюгирована со стрептавидином (см. рис. 4-58, Б). Чтобы выявить участок связывания зонда, препарат обрабатывают перекисью водорода и окрашивают; при этом пероксидаза обнаруживается в виде темной полосы на препарате (стрелка). (С любезного разрешения Tod Leverty, Gerald Rubin.)
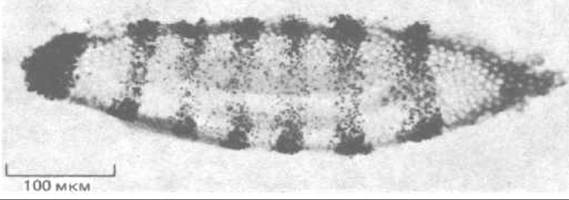
Рис. 4-75. Радиоавтограф среза очень раннего эмбриона Drosophila, подвергнутого гибридизации in situ с использованием радиоактивного ДНК-зонда, который комплементарен гену, принимающему участие в формировании сегментов ftz. Зонд гибридизуется с РНК эмбриона, и расположение зерен серебра после проявления указывает на то, что РНК, синтезируемая геном ftz, локализована в регулярных полосах, тянущихся поперек всего эмбриона. Ширина полос соответствует трем-четырем клеткам. На этих стадиях развития (клеточная бластодерма) эмбрион состоит примерно из 6000 клеток. (Из Е. Hafen, A. Kuroiwa, W. J. Gehring. Cell, 37; 833-841, 1984.)
4.6.13. Функция генов наиболее ярко проявляется в организме мутантов [49]
Предположим, что кому-то удалось клонировать ген, кодирующий вновь открытый белок. Как установить внутриклеточную функцию данного белка? Задача эта весьма непроста, поскольку ни трехмерная структура белка, ни полная нуклеотидная последовательность гена этого белка не позволяют судить о его функции. Кроме того, многие белки, например, структурные белки и белки сложных мультиферментных комплексов, будучи отделены от других компонентов сложной функциональной единицы, в состав которой они входят, не проявляют своей обычной активности.
Один из подходов, которые мы уже рассматривали (см. разд. 4.5.6) состоит в том, чтобы инактивировать определенный белок с помощью специфических антител и пронаблюдать за тем, какие клетки затронуты вследствие этого. В некоторых случаях такой способ позволяет достаточно надежно определить функцию белка, однако в отношении внутриклеточных белков он не очень эффективен, поскольку микроинъецированные антитела подвергаются разведению в процессе пролиферации клеток либо разрушаются в результате внутриклеточной деградации. Более удачное решение этой проблемы возможно с помощью генетических подходов. Мутанты, у которых отсутствует один из белков или, что более удобно, синтезируется его температурочувствительная форма (инактивируемая при небольшом повышении или понижении температуры), чрезвычайно полезны, когда перед исследователем стоит вопрос о функции белка. С их помощью идентифицированы функции ферментов, участвующих в основных метаболических путях бактерий. Благодаря этому подходу были открыты многие генопродукты, отвечающие за упорядоченное развитие эмбрионов Drosophila. Как правило, этот метод используется для изучения организмов с коротким циклом репродукции, таких, как бактерии, дрожжи, круглые черви и плодовые мушки. Воздействуя на их организм веществами, вызывающими изменения в ДНК (мутагенами), можно быстро получить большое количество мутантов и выбрать среди них тех, которые несут определенный интересующий экспериментатора дефект. Например, из популяции бактерий, подвергнутых воздействию мутагенов, были выбраны клетки, которые прекращают синтез ДНК при изменении температуры окружающей среды от 30° до 42° С. Среди них было выявлено значительное число температурочувствительных мутантов, мутации в которых затрагивали бактериальные белки, участвующие в процессе репликации ДНК. Эти мутанты позже были использованы для идентификации и определения белков, необходимых для репликации ДНК.
Цикл репродукции у человека очень продолжителен. К тому же никто не станет подвергать людей целенаправленному воздействию мутагенов. Более того, следует иметь в виду, что человеческий плод, имеющий серьезные дефекты жизненно важных процессов, например репликации ДНК, погибнет задолго до рождения. Однако многие мутации могут практически не оказывать влияния на жизнеспособность - например, тканеспецифические дефекты лизосом либо рецепторов клеточной поверхности, спонтанно возникающие в человеческой популяции. Анализ фенотипа этих больных, равно как и исследование их клеток в культуре, дают возможность уникального исследования важных клеточных функций. Хотя такие мутации крайне редки, тем не менее они эффективно выявляются, поскольку их носители обращаются за медицинской помощью.
4.6.14. Клетки и организмы, содержащие измененные гены, можно исправить [50]
Получить мутантов, у которых нарушена репликация ДНК или, например, развитие глаза, в принципе довольно просто. Однако, чтобы связать этот дефект с изменением конкретного белка, могут понадобиться годы. Технология рекомбинантных ДНК дала в руки исследователей совершенно иной подход: анализ начинается с белка и завершается созданием мутантной клетки или целого организма. Поскольку такой подход по сравнению с традиционным направлением генетического анализа от гена к белку представляется обратным, его обычно называют обратной генетикой.
Обратная генетика начинается с выделения из клетки нужного белка. Используя методы, описанные в гл. 5, ген этого белка клонируют и определяют его нуклеотидную последовательность; затем эту последовательность меняют биохимическими методами, создавая мутантный ген, кодирующий измененную форму белка. Затем такой ген вводят в клетку, где он может встроиться в хромосому в процессе гомологической рекомбинации и превратиться таким образом в постоянный элемент генома. Если встроенный ген экспрессируется, то несущая его клетка и все ее потомки будут синтезировать измененный белок. В том случае, когда измененный in vitro ген вводят в оплодотворенную яйцеклетку, получается многоклеточный мутантный организм. Некоторые из таких трансгенных организмов передадут этот ген своим потомкам в качестве постоянного элемента клеток зародышевой линии (рис. 4-76). Такая генетическая трансформация в настоящее время становится обычной процедурой для плодовых мушек или млекопитающих. В принципе на сегодняшний день совершенно реальна и трансформация человека, но такие эксперименты не выполняют из страха перед возможными генетическими нарушениями, которые нельзя исключить у лиц, ставших объектом таких исследований.
4.6.15. С помощью искусственных генов, кодирующих антисмысловые РНК, можно создавать специфические доминантные мутации [51]
При введении мутантных генов в клетки бактерий или дрожжей, которые, как правило, гаплоидны, такие гены будут достаточно часто рекомбинировать с нормальными гомологами. В результате можно отобрать клетки, в которых мутантный ген заменил единственную копию нормального гена (рис. 4-77, А), например клетки, синтезирующие определенный белок в мутантной форме. Функции нормального белка можно определить, как правило, по фенотипу мутантных клеток. Что же касается высших эукариот, таких, как млекопитающие или плодовые мушки, пока еще не созданы методы, позволяющие легко заменять нормальный ген клонированным мутантным геном. Генетическая трансформация таких организмов приводит обычно к инсерции клонированного гена в случайные участки генома и при этом клетка или организм содержат мутантный ген наряду с его нормальной копией (рис. 4-77, Б).

Рис. 4-76. Сравнение нормальной личинки Drosophila и двух мутантных личинок, содержащих дефектные гены ftz. Одна из дефектных (ftz') личинок была трансформирована после инъекции в яйцеклетку, из которой она была получена клонированной ДНК, содержащей нормальную последовательность гена ftz. Эта дополнительная последовательность ДНК встроилась в одну из хромосом мухи и затем нормально наследовалась и экспрессировалась. Ген ftz необходим для нормального развития и его добавление к дефектному геному, как следует из опыта, восстанавливает сегменты личинки, отсутствующие у организмов ftz'. Методы получения трансгенных животных обсуждаются далее (см. рис. 5-88). (С любезного разрешения Walfet Gehring.)

Рис. 4-77. Ген с измененной нуклеотидной последовательностью может быть введен в хромосому организма-хозяина. У бактерий и дрожжей можно отобрать мутанты, у которых (А) в результате генетической рекомбинации измененный ген занял место нормального. В этом случае в клетках сохраняются только мутантные гены. У высших эукариот вместо замены происходит добавление гена (Б). Трансформированные клетки или организмы у них содержат помимо нормальных мутантные гены. Полагают, что у организмов, для которых характерен избыток ДНК, замены генов происходят достаточно редко, поскольку для этого необходимо, чтобы мутантный ген «нашел» среди множества других последовательностей свой нормальный гомолог и спарился с ним.
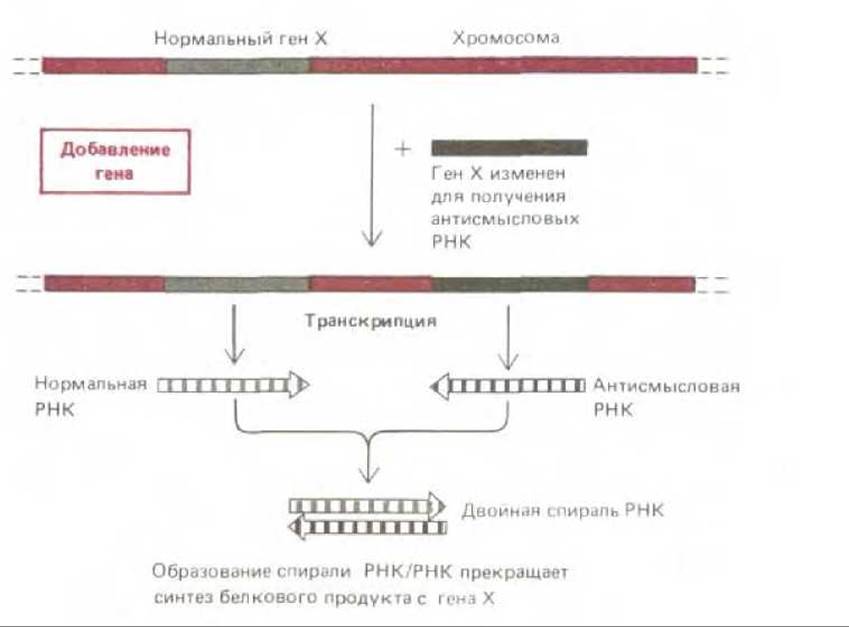
Рис. 4-78. Использование стратегии антисмысловых РНК для получения доминантных мутаций. Были сконструированы мутантные гены, синтезирующие РНК, последовательность которых комплементарна РНК, синтезируемым нормальными генами. РНК этих двух типов способны объединяться в двухцепочечные молекулы. Если синтезируется значительный избыток антисмысловых РНК, они могут гибридизоваться и таким образом инактивировать большую часть нормальных РНК, синтезируемых геном X. Полагают, что таким образом в перспективе удастся инактивировать любой ген. В настоящее время эта методика применима лишь по отношению к некоторым генам.
Было бы крайне полезно создание специфических доминантных мутаций в клетках высших эукариот путем введения мутантных генов, которые бы элиминировали активность их нормальных аналогов в клетке. Для этого используют весьма хитроумный и многообещающий подход, основанный на специфичности реакций гибридизации двух комплементарных цепей ДНК. Известно, что в норме только одна из двух цепей ДНК в данном участке транскрибируется в РНК и это всегда одна и та же цепь для данного гена. Если же клонированный ген был сконструирован таким образом, что транскрибируется только противоположно направленная цепь ДНК, появляется антисмысловая РНК с последовательностью, комплементарной нормальным гранскриптам РНК. В том случае, когда такая антисмысловая РНК синтезируется в достаточно больших количествах, она будет с большой частотой гибридизоваться со «смысловой» РНК, синтезируемой нормальными генами, и ингибировать синтез соответствующего белка (рис. 4-78). И если этот белок является жизненно важным для клетки или организма, описанные здесь доминантные мутанты погибнут и исследовать функции белка будет невозможно. Чтобы этого не произошло, можно сконструировать гены, синтезирующие антисмысловую РНК по команде, например, в ответ на изменение температуры или в присутствии определенной сигнальной молекулы. Клетки или организмы, содержащие такие индуцибельные антисмысловые гены, будут лишены специфического белка в определенное время, и в этом случае можно проследить за возникающим эффектом. Конечно, этот метод технически до конца еще не проработан, однако уже сейчас ясно, что он весьма перспективен для определения функции белков высших организмов.
Заключение
Технология рекомбинантных ДНК произвела революцию в исследовании клетки. В настоящее время с помощью рестрицирующих нуклеаз можно вырезать любой участок клеточной ДНК и вставить его в самореплицирующийся генетический элемент (плазмиду или вирус) для получения «клона геномной ДНК». С другой стороны, для получения «клона к ДНК» можно использовать ДНК-копию любой молекулы РНК. Таким образом, можно получить неограниченное количество высокоочищенной ДНК, определить ее нуклеотидную последовательность (скорость этой операции составляет сотни нуклеотидов в день), а также аминокислотную последовательность кодируемого ею белка. Методы генной инженерии позволяют синтезировать мутантные гены и вводить их в хромосомы клеток, где они в последующем превращаются в постоянный элемент генома. И если для переноса генов в качестве реципиента использовать оплодотворенное яйцо, могут быть получены трансгенные организмы, экспрессирующие мутантный ген и передающие его своим потомкам. Для клеточной биологии исключительно важно, что описанные методы дают возможность изменять клетки строго направленным образом, что в свою очередь позволяет оценить влияние на клетку изменения структуры определенного белка.
Применение технологии рекомбинантных ДНК открывает широкие перспективы. С помощью этих методов клетки бактерий, дрожжей и млекопитающих могут быть преобразованы в «фабрики» для масштабного производства любого белка. Это дает возможность детально анализировать структуру и функции белков или использовать их в качестве лекарственных средств. Кроме того, на технологии рекомбинантных ДНК основано получение высокоспецифических ДНК-зондов, с помощью которых изучают экспрессию генов в тканях, локализацию генов в хромосомах, выявляют гены, обладающие родственными функциями.
Литература
Общая
Cantor С. R., Schimmel P. R. Biophysical Chemistry, 3 vols. New York, W. H. Freeman, 1980 (A comprehensive account of the physical principles underlying many biochemical and biophysical techniques.)
Freifelder D. Physical biochemistry, 2nd ed. New York, W. H. Freeman, 1982.
Van Holde K.E., Physical Biochemistry, 2nd ed. Englewood, NJ, Prentice-Hall, 1985.
Цитируемая
1. Bradbury S. An Introduction to the Optical Microscope. Oxford, U. K., Oxford University Press, 1984.
FawcettD.W. A Textbook of Histology, llth ed. Philadelphia, Saunders, 1986.
2. SpencerM. Fundamentals of Light Microscopy. Cambridge, U. K., Cambridge University Press, 1982.
3. BoonM. Т., Drijver J. S. Routine Cytological Staining Methods. London, Macmillan, 1986.
4. Ploem J. S., Tanke H. J. Introduction to Fluorescence Microscopy. Royal Microscopical Society Microscopy Handbook No. 10. Oxford, U. K., Oxford Scientific Publications, 1987.
Willingham M. C., Pastan I. An Atlas of Immunofluorescence in Cultured Cells. Orlando, Fl, Academic, 1985.
5. Alien R. D. New observations of cell architecture and dynamics by video-enhanced contrast optical microscopy. Annu. Rev. Biophys. Biophys. Chem. 14, 265-290, 1985.
6. Pease D. C., Porter K. R. Electron microscopy and ultramicrotomy. J. Cell Biol., 91, 287s-291s, 1981.
7. Wischnitzer S. Introduction to Electron Microscopy. 3rd ed. Elmsford, NY, Pergammon, 1981.
8. Everhart Т.Е., Hayes T.L. The scanning electron microscope. Sci. Am., 226(1), 54-69, 1972. Hayat M. A. Introduction to Biological Scanning Electron Microscopy. Baltimore, University Park Press, 1978.
Kessel R.G., Kargon R.H. Tissues and Organs. New Fork, W. H. Freeman, 1979.
(Атлас тканей позвоночных, исследованных с помощью сканирующего электронного микроскопа).
9. Sommerville J., Scheer U. eds. Electron Microscopy in Molecular Biology. A Practical Approach. Washington, D. C, IRL Press, 1987.
10. Heuser J. Quick-freeze, deep-etching preparation of samples for 3-D electron microscopy. Trends Biochem. Sci, 6, 64-68, 1981.
Pinto da Silva P., Branton D. Membrane splitting in freeze-etching. J. Cell Biol, 45, 598-605, 1970.
11. Chiu W. Electron microscopy of frozen, hydrated specimens. Annu. Rev. Biophys. Biophys. Chem., 15, 237-257, 1986.
Unwin P. N. Т., Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J. Mol. Biol., 94, 425-440, 1975.
12. Glusker J.P., Trueblood K.N. Crystale Structure Analysis: A Primer. Oxford, U. K., Oxford University Press, 1985.
13. Alzari P.M., Lascombe M.B., Poljak R.J. Three-dimentional structure of antibodies. Annu. Rev. Immunol., 6, 555-580, 1980.
Kendrew J.C. The three-dimentional structure of protein molecule. Sci. Am., 205(6), 96-111, 1961. Perutz M.F. The hemoglobin molecule. Sci. Am., 211(5), 64-76, 1964.
14. Cooke R.M., Cambell I.D. Protein structure determination by NMR. Bioessays, 8, 52-56, 1988.
Schulman R.G. NMR spectroscopy of living cells. Sci. Am., 248(1), 86-93, 1986. Wuthrich K., Wagner G. Internal dynamics of proteins. Trends Biochem. Sci., 9, 152-154, 1984.
15. AmmanD. Ion Selective Microelectrodes: Principles, Design and Application. Berlin, Springer-Verlag, 1986.
Auerbach A., Sachs F. Patch clamp studies on single ionic channels. Annu Rev. Biophys. Bioeng., 13, 269-302, 1984.
16. Grynkiewicz G., Poenie M., Tsien R. Y. A new generations of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem, 260, 3440-3450, 1985. Tsien r. Y., Poenie M. Fluorescence ratio imaging: a new window into intracellular ionic signalling. Trends Biochem. Sci., 11, 450, 1986.
17. Celis J.E., Graessmann A., Logter A. eds. Microinjection and Organdie Transplantation Techniques. London, Academic Press, 1986.
Gomperts B. D., Fernandez J. M. Techniques for membrane permeabilization. Trends Biochem. Sci., 10, 414-417, 1985.
Ostro M.J. Liposomes Sci. Am., 256(1), 102-111, 1987.
Ureta Т., Radojkovic J. Microinjected frog oocytes. Bioessays, 2, 221-225, 1985.
18. FreshneyR.I. Culture of Animal Cells: A Manual of Basic Technique. New York, Liss, 1987.
19. HerzenbergL. A. SweetR.G., HerzenbergL. A. Fluorescence-activated cell sorting. Sci. Am., 234(3), 108-116, 1976.
KamarckM. E. Fluorescence-activated cell sorting of hybrid and transfected cells. Methods Enzymol, 151, 150-165, 1987.
Nolan G.P., Fiering S., Nicolas J.F., Herzenberg L.A. Fluorescence-activated cell analysis and sorting of viable mammalian cells based on beta-D-galactosidase activity after transduction ofE. coli lac Z. Proc. Nat. Acad. Sci. USA, 85, 2603-2607, 1988.
20. Harrison R.G. The outgrowth of the nerve fiber as a mode of protoplasmic movement. J. Exp. Zool., 9, 787-848, 1910.
21. Ham R.G. Clonal growth of mammalian cells in a chemically defined, synthetic medium. PNAS, 53, 288-293, 1965.
LooD. Т., Fuquay J. І., Rawson C. L., BarnesD. W. Extended culture of mouse embryo cells without senescence: inhibition by serum. Science, 236, 200-202, 1987.
Sirabasku D.A., Pardee А. В., Saw G.H., eds. Growth of Cells in Hormonally Defined Media. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1982.
22. RuddleF. H., CreaganR. P. Parasexual approaches to the genetics of man. Annu. Rev. Genet., 9, 407-486, 1975.
23. Colowick S. P., Kaplin N. O., eds. Methods in Enzymology, vols. 1 -... San Diego, CA, Academic Press, 1955-1988. (Многотомное издание, содержащее общие и специальные статьи с описанием множества методов).
Cooper T.G. The Tools of Biochemistry. New York, Wiley, 1977.
Scopes R.K. Protein Purification Principles and Practice, 2nd ed. New York, Springer-Verlag, 1987.
24. Claude A. A coming age of cell. Science, 189, 433-435, 1975. de Duve C., Beaufay H. A short history of tissue fractionation. J. Cell Biol., 91, 293s-299s, 1981.
Meselson M., Stahl F. W. The replication of DNA in Escherichia coli. Proc. Nat. Acad. Sci. USA, 47, 671-682, 1958. (Для демонстрации полуконсервативной репликации ДНК использовали центрифугирование в градиенте плотности).
Palade G. Intracellular aspects of the process of protein synthesis. Science, 189, 347-358, 1975.
Sheeler P. Centrifugation in Biology and Medical Science. New York, Wiley, 1981.
25. Moore D.J., Howell K.E., Cook GM. W., Ewans WH. eds. Cell Free Analysis of Membrane Traffic. New York, Liss, 1986.
Nirenberg N. W., Mattaei J. H. The dependence of cell free protein synthesis in E. coli on naturally occuring or synthetic polyribonucleotides. Proc. Nat. Acad. Sci. USA, 47, 1588-1602, 1961.
Racker E. A. A New Look on Mechanisms of Bioenergetics. New York. Academic Press, 1976. (Бесклеточные системы в исследовании энергетического метаболизма).
Zamecnic P. С. An historical account of protein synthesis with current overtones - a personalized view. Cold Spring Harbor Symp. Quant. Biol., 34, 1-16, 1969.
26. DeanP. D. G., Johnson W. S., MiddleF. A. Affinity Chromatography: A Practical Approach, Arlington, VA, IRL Press, 1985.
GilbertM. T. High Performance Liquid Chromatography. Littleton, MA, John Wright-PSG, 1987.
27. Andrews A. T. Electrophoresis, 2nd ed. Oxford, U. K., Clarendon Press, 1986.
Laemmli U. K. Cleavage of the structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680-685, 1970.
28. Celis J. E., Bravo R. eds., Two - Dimentional Gel Electrophoresis of Proteins. New York, Academic Press, 1983.
O'FarrellP.H. High-resolution two-dimentional electrophoresis of proteins. J. Biol. Chem. 250, 4007-4021, 1975.
29. Cleveland D. W., Fisher S. G., Kirschner M. W., Laemmli U. K. Peptide mapping by limited proteolysis in sodium dodecyl sulphate and analysis by gel-electrophoresis. J. Biol. Chem. 252, 1102-1106, 1977.
Ingram К М. A special chemical difference between the globins of normal human and sickle anemia hemoglobin. Nature, 178, 792-794, 1956.
30. Edman P., Begg G. A protein sequenator. Eur. J. Biochem. 1, 80-91, 1967. (Первое описание автоматизированного секвенатора).
Hewlck R. M., Hunkapiller M. W., Hood L. E., Dreyer WJ. A gas-liguid-solid phase peptide and protein seguenator. J. Biol. Chem. 256, 7990-7997, 1981.
Sanger F. The arrangement of amino acids in proteins. Adv. Protein Chem. 7,1 -67, 1952.
Walsh K. A., Ericsson L. H. Parmelee D. C., Titani K. Advances in protein sequencing. Annu. Rev. Biochem. 50, 261-284, 1984.
31. Chase G.D., Rabinowltz J.L. Principles of Radio Isotope Methodology, 2nd ed. Minneapolis, Burgess, 1962.
Dyson N. A. An Introduction to Nuclear Physics with Applications in Medicine and Biology, Chichester, U. K., Horwood, 1981.
32. Calvin M. The path of carbon in photosynthesis. Science, 135, 879-889, 1962. (Один из первых случаев применения радиоизотопов в биологии).
Rogers A. W. Techniques of Autoradiography, 3rd ed. New York, Elsevier/North Holland, 1979.
33. Anderton B. H., Thorpe R. C. New methods of analysis of antigens and glycoproteins in complex mixtures. Immunol. Today, 2, 122-127, 1980.
Coons A.H. Histochemistry with labelled antibody. Int. Rev. Cytol. 5, 1-23, 1956.
34. Milstein C. Monoclonal antibodies. Sci. Am., 243(4), 66-74, 1980.
YeltonD. E., Schariff M. D. Monoclonal antibodies: a powerful new tool in biology and medicine. Annu. Rev. Biochem., 50, 657-680, 1981.
35. Mabuchi L, Okuno M. The effect of myosin antibody on the division of starfish blastomeres. J. Cell Biol., 74, 251-263, 1977.
Mulcahy L. S., Smith M. R., Stacey D. W. Requirement for ras proto-oncogene function during serum stimulated growth of NIH 3T3 cells. Nature, 313, 241-243, 1985.
36. Drlica K. Understanding DNA and Gene Cloning. New York, Wiley, 1984.
Sambrook J., Fritsch E. F., Maniatis T. Molecular Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 1989.
Watson J. D., Tooze J. The DNA Story: A Documentary History of Gene Cloning. New York, W.H. Freeman, 1981.
37. Garoff H. Using recombinant DNA techniques to study protein targeting in the eucaryotic cell. Annu. Rev. Cell. Biol., 1, 403-445, 1985.
Jackson lan J. The real reverse genetics: targeted mutagenesis in the mouse. Trends Genet., 3, 119, 1987.
Kelly J.H., Darlington G.J. Hybrid genes: molecular appoaches to the tissue-specific gene regulation. Annu. Rev. Genet., 19, 273-296, 1985.
38. Nathans D., Smith H. 0. Restriction nucleases in the analysis and reconstructuring of DNA molecules. Annu. Rev. Biochem., 44, 273-293, 1975.
Smith H. 0. Nucleotide sequence specificity of restriction endonucleases. Science, 205, 455-462, 1979.
39. Cohen S.N. The manipulation of genes. Sci. Am., 233(1), 24-33, 1975.
Maniatis T. et al. The isolation of structural genes from libraries of eucaryotic DNA. Cell, 15, 687-701, 1978.
Nooick R.P. Plasmids. Sci. Am., 243(6), 102-127, 1980.
40. Cantor C. R., Smoth C. L., Mathew M. K. Pulsed-field gel electrophoresis of very large DNA molecules. Annu. Rev. Biophys. Biophys. Chem., 17, 287-304, 1988.
Maxam A. M., Gilbert W, A new method of sequencing DNA. Proc. Nat. Acad. Sci. USA, 74, 560-564, 1977.
Southern E. M. Gel electrophoresis of restriction fragments. Methods Enzymol., 68, 152-176, 1979.
41. Rigby P. W., Dieckermann M., Rhodes C., Berg P. Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase I. J. Мої. Biol, 113, 237-251, 1977.
42. Galas D. J., Schmitz A. DNAse footprinting: a simple method for the detection of protein-DNA binding specificity. Nucl. Acid Res., 5, 3157-3170, 1978.
Gilbert W. DNA sequencing and gene structure. Science, 214, 1305-1312, 1981.
Prober J. M., et al. A system for rapid DNA sequencing with fluorescent chainterminating dideoxynucleotides. Science, 238, 336-341, 1987. Tullius T. D. Chemical "snapshots" of DNA: using the hydroxyl radical to study the structure of DNA and DNA protein complexes. Trends Biochem. Sci., 12, 297-300, 1987.
43. Berk A.J., Sharp P. A. Sizing and mapping of early adenovirus mRNAs by gel electrophoresis of SI endonuclease digested hybrids. Cell, 12, 721-732, 1977.
Hood L.E., Wilson J.H., Wood W.B. Molecular Biology of Eucaryotic Cells: A Problems Approach, pp. 56-61, 192-210, Mento Park, CA, Benjamin-Cummings, 1975.
Wetmer J. G. Hybridization and renaturation kinetics of nucleic acids. Annu. Rev. Biophys. Bioeng., 5, 337-361, 1976.
44. Alwine J. C., Kemp D. J., Stark G. R. A method for detection of specific RNAs in agarose gels by transfer to diazobenzomethyl-paper and hybridization with DNA probes. Proc. Nat. Acad. Sci. USA. 74, 5350-5354, 1977.
Southern Е. М. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Мої. Biol., 98, 503-517, 1975.
45. Itakura K., Rossi J. J., Wallace R. B. Synthesis and use of synthetic oligonucleotides. Annu. Rev. Biochem., 53, 323-356, 1984.
Ruddle F. H. A new era in mammalian gene mapping: somatic cell genetics and recombinant DNA methodologies. Nature, 294, 115-119, 1981.
White R., Lalovel J. M. Chromosome mapping with DNA markers. Sci. Am., 258(2), 40-48, 1988.
McGinnis W., Garber R.L., Wirz J., Kuroiwa A., Gehring WJ. A homologous protein-coding seguence in Drosophila homeotic genes and its conservation in other metazoans. Cell, 37, 403-408, 1984.
47. GerhardD. S., Kawasaki E. S., Bancroft F. C., Shabo P. Localization of a unique gene by direct hybridization. Proc. Nat. Acad. Sci. USA, 78, 3755-3759, 1981.
Huten E., Kuroiwa A., Gehring W.J. Spatial distribution of transcripts from the segmentation gene fushi tarazu during Drosophila embryonic development. Cell, 37, 833-841, 1984.
Pardue M. L., Gall J. G. Molecular hybridization of radioactive DNA to the DNA of the cytological preparations. Proc. Nat. Acad. Sci. USA, 64, 600-604, 1969.
48. Abelson J., ButzE., eds. Recombinant DNA. Science, 209, 1317-1438, 1980.
Gilbert W., Villa-Komaroff L. Useful proteins from recombinant bacteria. Sci. Am., 242(4), 74-94, 1980.
49. Lederberg J., Lederberg E. M. Replica plating and inderect selection of bacterial mutants. J. Bacteriol., 63, 399-406, 1952.
Nusslein-Volhard C., Wieschaus E. Mutations affecting segment number arid polarity in Drosophila. Nature, 287, 795-801, 1980.
50. Palmiter R. D., BrinsterR. L. Germ line transformation of mice. Annu. Rev. Genet., 20, 465-499, 1986.
Pellicer A., et al. Altering genotype and phenotype by DNA-mediated gene transfer. Science, 209, 1412-1422, 1980.
Rubin G. M., Spradling A. C. Genetic transformation of Drosophila with transposable element vectors. Science, 218, 348-353, 1982.
Shortle D., Nathans D. Local mutagenesis: a method for generating viral mutants with base substitutions in preselected regions of the viral genome. Proc. Nat. Acad. sci. USA, 75, 2170-2174, 1978.
StruhlK. The new yeast genetics. Nature, 305, 391-397, 1983.
51. Melton D.A., Rebagliati M.R. Antisense RNA injections in fertilized eggs as a test for the function of localized mRNAs. J. Embryol. Exp. Morphol., 97, 211-221, 1986.
Rosenberg V.B., Preiss A., Seifert E., Jackle H., Knipple D.C. Production of phenocopies by Kruppel anti-sense RNA injection into Drosophila embryos. Nature, 313, 703-706, 1985.
Weintraub H., Isant J. G., HarlandR. M. Anti-sense RNA as a molecular tool for genetic analysis. Trends Genet., 1, 22-25, 1985.