Молекулярная биология: Структура и функции белков - Степанов В.М. 2005
Ферменты
Сериновые протеиназы
Сериновые протеиназы, названные так по аминокислотному остатку, характерному для их активных центров, широко распространены в природе и вместе с протеолитическими ферментами других классов (аспартилъными, цистеиновыми и металлопротеиназами) обеспечивают глубокое расщепление белков — основу их катаболизма — и целый ряд реакций ограниченного протеолиза, имеющих регуляторное значение. Известно несколько семейств, объединяющих эволюционно родственные сериновые протеиназы. Для животного мира очень характерны многообразные сериновые протеиназы, принадлежащие к семейству химотрипсина, т.е сходные с химотрипсином (наиболее изученным ферментом этого типа) по пространственной структуре, строению и механизму действия каталитического центра. К этому же семейству относятся некоторые сериновые протеиназы стрептомицетов. Сериновые протеиназы другого семейства — субтилизины — особенно характерны для прокариот, но встречаются также в растениях и животных. Известно и третье семейство — сериновые карбоксипептидазы, продуцируемые животными, грибами, дрожжами и растениями. Однотипность строения каталитических центров и глубокое сходство механизмов действия ферментов этих семейств, совершенно не похожих друг на друга по пространственной структуре, объясняются, по-видимому, конвергентной эволюцией белков-предшественников.
В дальнейшем изложении обобщены данные о механизме действия сериновых протеиназ.
10.5.1. Связывание субстрата
Зона связывания субстрата в протеиназах, участвующая в образовании комплексов с белками или их фрагментами, достаточно протяженна. Обычно с ней могут взаимодействовать 5-8 аминокислотных остатков — по 2 4 по обе стороны расщепляемой пептидной связи. Наблюдать связывание истинных субстратов — пептидов значительной длины — методами рентгеноструктурного анализа не удается из-за чрезвычайно малых времен существования таких комплексов, в которых субстрат быстро расщепляется. Ввиду этого исследуют комплексы ферментов с аналогами субстратов. Так, подробно описано взаимодействие альдегидопептидов (соединений, в которых С-концевой остаток заменен аминоальдегидом) с ферментом семейства химотрипсина — протеиназой А стрептомицетов:

При взаимодействии с сериновой протеиназой альдегидная группа образует полуацеталь с гидроксилом серина активного центра. Эта связь служит моделью переходного фермент-субстратного комплекса, получающегося при взаимодействии с гидроксильной группой остатка серина в активном центре и карбонила расщепляемой пептидной связи:

Образующийся весьма стабильный комплекс позволяет описать систему взаимодействий между ферментом и субстратом на участке, предшествующем атакуемой пептидной связи. Взаимодействия фермента и С-концевой части субстрата изучали на других моделях, в частности на комплексах ферментов и природных белковых ингибиторов протеиназ.
На участке, предшествующем атакуемой пептидной связи, фермент формирует с субстратом своего рода антипараллельную ß-структуру, образуя три водородные связи. Их дополняют еще четыре водородные связи с функциональными группами каталитического центра. Укладывание пептидного субстрата в зоне связывания сопровождается небольшим смещением окружающих ее участков фермента, а также отщеплением цепочки молекул воды, которые гидратировали функциональные группы свободного фермента, образуя с ними водородные связи.
Система водородных связей между субстратом и ферментом дополняется рядом гидрофобных контактов. Важнейшие из них образует боковая цепь того аминокислотного остатка, карбонильная группа которого принадлежит атакуемой ферментом пептидной связи. Согласно принятой номенклатуре, такой аминокислотный остаток обозначают Р1, предшествующий ему — Р2, далее Р3, Р4 и т.д., следуя к аминоконцевому остатку субстрата. Остаток, чья аминогруппа образует атакуемую связь, обозначают Р'1 следующий — Р'2и т.д Соответствующие этим остаткам участки зоны связывания субстрата обозначают S4, S3, S2, S1, S'1, S'2.
Протеиназа А стрептомицетов, гидролизующая пептидные связи, в которых участвует карбоксильная группа гидрофобных аминокислот, т.е. близкая по специфичности к химотрипсину, образует с боковой цепью остатка Р1 около 40 нековалентиых контактов. Связывание этой гидрофобной группы сопровождается вытеснением молекул воды из гидрофобной впадины S1. Столь развитая система нековалентных контактов придает особо важное значение остатку Р1, природа которого в основном определяет выбор расщепляемой пептидной связи сериновыми протеиназами, иными словами, специфичность этих ферментов, хотя последняя зависит (пусть в меньшей степени) и от соседних аминокислотных остатков.
Интересно, что в образовании гидрофобных контактов с субстратом участвуют остатки аланина, глицина, треонина, глутамина, пролина, выстилающие зону S1 фермента. На первый взгляд участие этих, в большинстве своем гидрофильных, аминокислот в построении гидрофобной зоны связывания парадоксально, однако в этом сказывается принципиальная разница в поведении одних и тех же остатков в растворе и при включении в фиксированную пространственную структуру белка. В последней даже метиленовая группа глицина, будучи как бы отделенной, изолированной от гидрофильных пептидных связей, способна вести себя как гидрофобный элемент. Такие небольшие сами по себе элементы, закрепленные в пространстве водородными связями соседних групп, образуют своего рода гидрофобную мозаику, выстилающую впадину, «карман» в зоне связывания субстрата.
Видимо, такой способ формирования гидрофобных зон даже эффективнее, чем использование для этой цели отчетливо гидрофобных аминокислот с крупными боковыми группами, поскольку зафиксировать последние в поверхностной структуре белка, в частности лишить их способности вращаться, сложнее, чем фиксировать маленький гидрофобный элемент.
У сериновых, протеиназ, специфически расщепляющих пептидные связи, которые образованы карбоксильными группами лизина или аргинина — трипсина и его аналогов, — также реализуются множественные контакты между полипептидной цепью субстрата и ферментом. И в этом случае особенно важно установление системы водородных связей между пептидной цепью субстрата и ферментом:
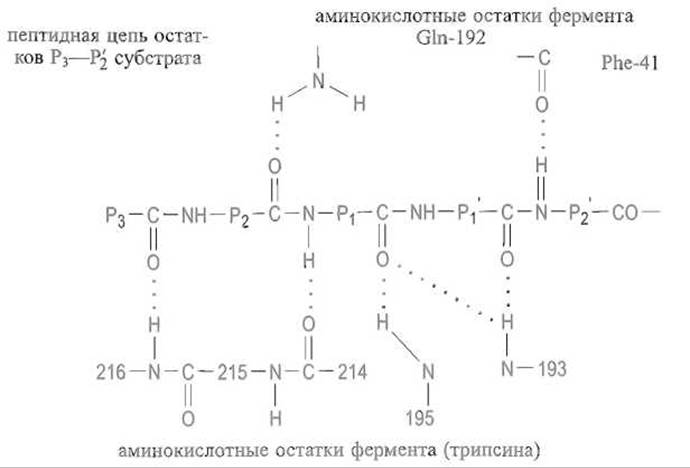
Связывание остатка Р1 в основном определяется электростатическими взаимодействиями между катионными группами в боковой цепи субстрата — NH+3 в случае лизина или гуанидиниевой группой
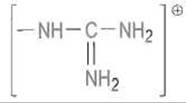
в случае аргинина — и карбоксилат-анионом остатка аспарагиновой кислоты, расположенного на дне гидрофобной впадины S1 (Asp-189 в трипсине) (рис. 10.3). Такому электростатическому взаимодействию благоприятствует изоляция ионной пары от контакта с водой.
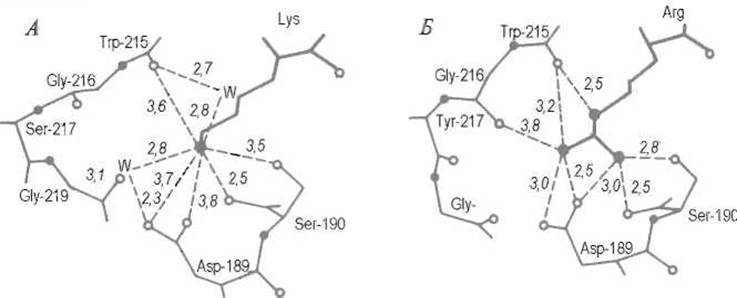
Рис. 10.3. Размещение боковой цепи остатка P1 субстрата в зоне S1 трипсина.
А — взаимодействия NH’3-группы лизина, Б - гуанидогруппы аргинина. Светлыми кружками обозначены атомы кислорода, темными — азота, W — молекулы воды, удерживаемые ферментом; пунктиром показаны водородные связи, цифры соответствуют их размерам в ангстремах
Кроме того, оно дополняется системой водородных связей между гуанидо- или аминогруппой субстрата и карбоксилат-ионом Asp-189, а также кислородными атомами ряда С=O-групп фермента. Замена остатка Asp-189 на остаток лизина в трипсине, проведенная методами белковой инженерии, радикально изменила специфичность фермента, который перестал гидролизовать пептидные связи аргинина и лизина, но приобрел (правда, слабую) способность расщеплять связи, образованные гидрофобными остатками фенилаланина, лейцина, тирозина, т.е. по специфичности стал близким другому ферменту того же семейства — химотрипсину.
Иногда остаток Р1 не столь доминирует в определении специфичности, и больший вес приобретают взаимодействия с ферментом других аминокислотных остатков субстрата. Например, специфичность калликреина — аналога трипсина, вовлеченного в регуляторные механизмы у животных, — определяется присутствием аргинина в положении P1 и гидрофобного остатка в положении P2 субстрата и оказывается, таким образом, гораздо более узкой.
При действии протеиназ на нативные белковые субстраты специфичность гидролиза определяется не только и даже не столько последовательностью аминокислот расщепляемого участка, сколько его пространственной, стерической доступностью. Так, погружение всего активного центра калликреина в своего рода впадину за счет удлинения прилегающих к нему пептидных петель фермента делает практически невозможным гидролиз обычных белковых субстратов, даже если они содержат последовательность гидрофобный остаток — аргинин. Калликреин атакует только такой белок, в котором соответствующая его специфичности последовательность расположена на выступе белка-субстрата, комплементарном впадине активного центра фермента. Так достигается очень узкая, практически уникальная специфичность калликреина — фермента, участвующего в отщеплении вазоактивных пептидов от белковых предшественников, но практически не действующего на другие белки.
Отметим еще одну особенность связывания белковых субстратов в активном центре протеиназ. При образовании комплексов с достаточно длинными пептидами, перекрывающими активный центр, укладка полипептидной цепи в зоне связывания требует, как предполагают, определенного ее «перекручивания», некоторого искажения плоскостной структуры атакуемой ферментом пептидной связи, что создает благоприятные условия для ее перехода из плоской в пирамидальную конфигурацию, близкую к переходному состоянию. Следовательно, уже на стадии образования комплекса Михаэлиса при связывании субстрата подготавливается стереохимический переход, имеющий ключевое значение для эффективного катализа.
Характерные для сериновых протеиназ черты связывания субстрата — множественность взаимодействий, точность ориентации относительно групп каталитического центра и преимущественное связывание конформации, стереохимически: приближенной к той, которая существует в переходном комплексе, — типичны для многих ферментов. Необходимо подчеркнуть, что две важнейшие стадии катализа — связывание субстрата и собственно каталитическое превращение — хотя и рассматриваются раздельно, в действительности слиты в единый механизм.
10.5.2. Каталитический механизм
Топография каталитического центра сериновых протеиназ описана достаточно подробно. Изучение структуры комплексов ферментов этого класса с различными аналогами субстратов и ингибиторами в сочетании с данными по кинетике катализа, химической модификации отдельных групп фермента, метода ЯМР и белковой инженерии привели к следующим представлениям о механизме действия сериновых протеиназ.
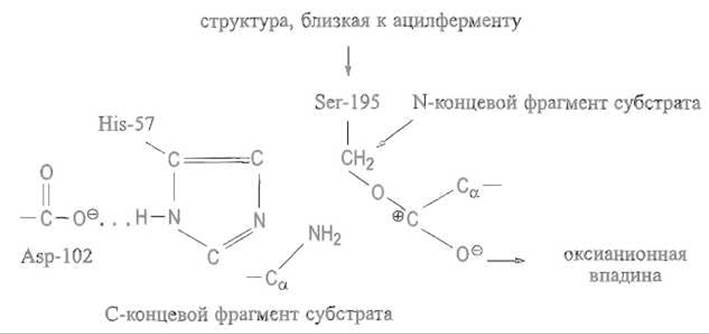
В превращении субстратов наибольшая роль принадлежит гидроксильной группе серина Ser-195 (здесь и далее нумерация остатков дается по последовательности наиболее изученного фермента — а-химотрипсина), имидазолу His-57, ß-карбоксильной группе Asp-102, а также пептидным группам остатков Ser-195 и Gly-193. Таким образом, в сериновых протеиназах, как и в других ферментах, активный центр собран из аминокислотных остатков, расположенных в совершенно разных участках полипептидной цепи, но сближенных в пространственной структуре фермента (рис. 10.4).
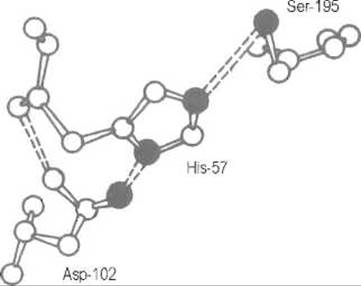
Рис. 10.4. Расположение функциональных групп в каталитическом центре сериновых протеиназ.
Показаны компоненты каталитического центра — остатки серина, гистидина и аспарагиновой кислоты; зачернены атомы азота и кислорода, участвующие в механизме катализа. Прерывистыми линиями обозначены водородные связи. Нумерация дана по аминокислотной последовательности химотрипсина; в других сериновых протеиназах эти аминокислоты могут занимать другие места в первичной структуре фермента, но их относительное расположение в пространстве строго сохраняется
Как правило, компоненты каталитического центра взаимодействуют с соседними группами фермента, что закрепляет их в определенной ориентации и видоизменяет химические характеристики.Например, карбоксильная группа остатка Asp-102 образует с соседями четыре водородные связи, которые стабилизируют ее анионную форму и как следствие смещают рКа этого остатка к 2-3 вместо 3,6 (величине, характерной для обычной ß-карбоксильной группы аспарагиновой кислоты). Соседство этого карбоксилат-иона стабилизирует протон на одном из атомов имидазольной группы гистидина — именно на том, который обращен к Asp-102, — и вместе с тем определенную электронную структуру имидазольного кольца His-57. Последнее очень характерно для функциональных групп белка и не имеет аналогий в поведении таких же групп, в частности имидазольной, в небольших молекулах. Окружение функциональных групп в белке анизотропно.
Влияние микроокружения на поведение функциональных групп в ферменте весьма существенно, и все же реакционная способность компонентов активного центра сериновых протеиназ в отсутствие субстрата не обнаруживает резких различий, которые выходили бы за рамки обычных вариаций в химических свойствах функциональных групп на поверхности белка. Так, сообщения о том, что имидазол His-57 и карбоксил Asp-102 в активном центре белка как бы поменялись свойствами, причем имидазол оказался более кислым, чем карбоксильная группа, оказались ошибочными. Точно так же нельзя думать, что в свободном ферменте происходит эстафетный перенос отрицательного заряда от карбоксилат-иона Asp-102 на Ser-195 и гидроксил последнего превращается в гидроксидный ион — чрезвычайно сильный нуклеофил. Такая передача заряда была бы сразу же обесценена реакцией гидроксид-иона с водой:
![]()
Напротив, в фермент-субстратном комплексе химические свойства участников каталитического процесса, в том числе функциональных групп каталитического центра, меняются очень существенно, что определяется не только развитой системой нековалентных взаимодействий, охватывающей фермент-субстратный комплекс, но и изоляцией всей системы от воды. По справедливому замечанию М. Дьюара, само связывание фермента и субстрата предполагает как необходимую предпосылку отделение молекул воды, окружающих до реакции субстрат и заполняющих зону активного центра фермента. Вследствие этого каталитическая реакция переносится в совершенно иную среду. Это соображение еще раз подчеркивает неразрывную связь обеих сторон биокаталитического процесса — связывания и собственно превращения субстрата.
Гипотетический механизм действия сериновых протеиназ может быть описан следующим образом (рис. 10.5). Необходимо учитывать, что происходящие последовательно процессы в действительности могут протекать синхронно.
Связывание полипептидного субстрата в активном центре ставит карбонильную группу атакуемой пептидной связи в такую позицию, что ее кислород оказывается в так называемой оксианионной впадине. Образование этим атомом двух водородных связей с соответствующим образом ориентированными N—Н-группами, принадлежащими пептидным связям остатков Ser-195 и Gly-193, способствует поляризации связи С=O и ее переходу в С—О, стабилизирует оксианион. Положительный заряд на углероде карбонильной группы за счет точного связывания субстрата ферментом пространственно сближен с кислородом гидроксильной группы серина. Это вызывает поляризацию гидроксильной группы, которая принимает структуру близкую к —СН2—О- ... Н+, что резко усиливает нуклеофильность атома кислорода серина.
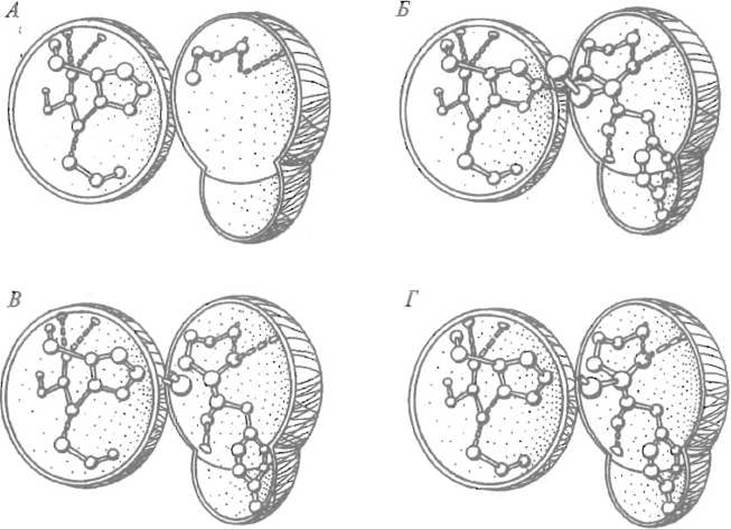
Рис. 10.5. Механизм действия сериновых протеиназ.
А — функциональные группы в активном центре фермента. В левой полусфере — имидазол гистидина, за которым расположен карбоксил остатка аспарагиновой кислоты, фиксированный системой водородных связей, в правой — боковая цепь серина и две водородные связи оксианионной впадины.
Б — в правой полусфере — продукт взаимодействия субстрата (показан тирозин в положении Р1); кислород размещен в оксианионной впадине; протон, покинувший кислород гидроксильной группы серина, перемещается к азоту имидазола и далее — к атому азота расщепляемой
пептидной связи.
В — молекула воды разместилась вблизи атома углерода карбонильной группы, причем ее протон переместился к азоту имидазола гистидина.
Г— гидроксильная группа воды присоединилась к углероду карбонильной группы, чем завершилось расщепление пептидной группы
Таким образом, в рассматриваемой схеме увеличение нуклеофильности серина происходит под влиянием соответственно ориентированного и поляризованного за счет взаимодействия с ферментом карбонила субстрата, причем последний экранирует нуклеофил, защищая его от воды.
Далее образуется ковалентная связь между углеродом субстрата и кислородом субстрата, вероятно, более длинная и менее прочная, чем обычно (рис. 10.5, Б):

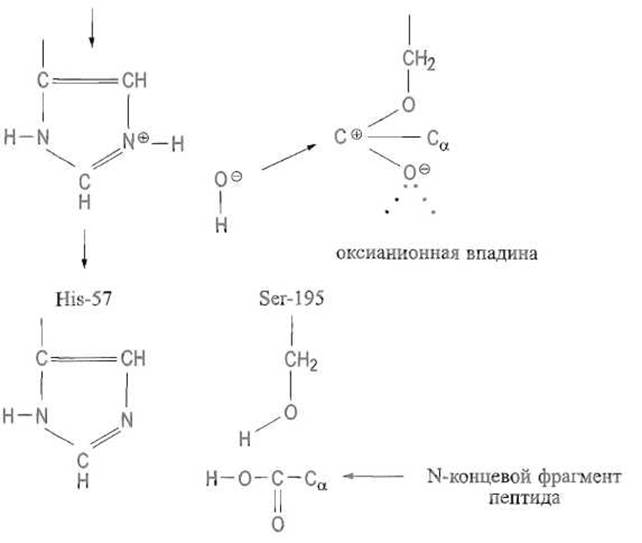
Протон, отщепившийся от гидроксила серина, перемещается по траектории, которая задана имидазольной группой His-57, приближаясь к непротонированному атому азота, а затем переходит к —NH-группе расщеплямой пептидной связи. Как известно, атом азота в пептидных группах практически лишен основных свойств из-за смещения свободной электронной пары, обслуживающей связь С—N, которая носит характер частично двойной. Однако после поляризации карбонильной группы в оксианионной впадине плоскостной характер пептидной связи утрачивается, связь С—N становится простой и атом азота приобретает свойства весьма сильного основания. Ввиду этого протон от имидазольной группы — слабого основания — переходит к группе — NH-, описав своего рода дугу. Связь С—N разрывается и фрагмент пептидного субстрата высвобождается (рис. 10.5, В).
Таким образом, выполнена первая задача катализа — расщеплена пептидная связь и высвобожден С-концевой фрагмент пептида. Оставшаяся часть пептида (от остатка P1 до аминного конца) пока сохраняет связь с ферментом через остаток серина. По-видимому, эта связь может в определенных условиях стабилизироваться за счет восстановления карбонильной группы, взаимной компенсации положительного и отрицательного зарядов, так что фрагмент пептида образует с ферментом сложноэфирную связь — образуется так называемый ацилфермент:

При действии сериновых протеиназ на некоторые аналоги субстратов удается наблюдать, а иногда и выделить достаточно устойчивый ацилфермент. При гидролизе обычных субстратов, однако, такая стабилизация, видимо, не обязательна, и гидролизу подвергается производное с поляризованной карбонильной группой. Во всяком случае, после ухода С-концевого фрагмента пептида молекула воды занимает освободившееся место и располагается так, что образует водородную связь с азотом имидазольной группы, тогда как ее атом кислорода оказывается сближенным с положительно заряженным атомом углерода карбонильной группы. Это приводит к отщеплению протона, присоединяющегося к имидазолу, и образованию гидроксильного иона, легко взаимодействующего с положительно заряженным углеродом карбонила. Протон мигрирует к кислороду серина, в результате чего с расщеплением связи С—О восстанавливается гидроксильная группа активного центра и высвобождается N-концевой фрагмент пептида, чем и завершается каталитический цикл (рис. 10.5, Г):

Эта стадия реакции протекает в целом аналогично первой, однако роль гидроксильной группы серина переходит к молекуле воды.
Если принять, что происходит стабилизация промежуточного продукта и образуется ацилфермент, механизм гидролиза последнего окажется практически таким же. Следует лишь предположить поляризацию С = О-связи в сложноэфирной группе ацилфермента за счет водородных: связей в оксианионной впадине, что приведет к появлению положительного заряда на атоме углерода карбонильной группы, т.е. к образованию того же самого промежуточного продукта, судьба которого прослежена выше.
Важнейшей чертой рассмотренного механизма действия сериновых протеиназ, по-видимому типичной и для других ферментов, является стабилизация тетраэдрического переходного состояния в фермент-субстратном комплексе. Характерно, что весьма эффективными ингибиторами сериновых протеиназ являются алкилборные кислоты, в которых предсуществует тетраэдрическая конфигурация заместителей — трех гидроксильных групп и алкильного остатка, — имитирующая структуру переходного состояния. Особенно эффективны производные боронатного аналога фенилаланина, который связывается в участке S1 за счет бензильной группы и в каталитическом центре; соответствующая константа ингибирования Ki составляет 10-8 М:

Альдегидопептиды — ингибиторы сериновых протеиназ, как упоминалось, образуют с серином активного центра ковалентную полуацетальную связь — структуру, аналогичную в известной мере ацилферменту. При этом свободный гидроксил связывается в оксианионной впадине так же, как это происходит с кислородом карбонильной группы в фермент-субстратном комплексе.
Хорошо известны ингибиторы сериновых протеиназ, которые при взаимодействии с ферментом проходят первую фазу каталитического процесса и образуют ацилфермент. Некоторые такие производные гидролизуются с измеримой скоростью, однако другие, в частности производные арилсульфофторидов и ряд фосфорорганических соединений, оказываются стабильными, что приводит к необратимому ингибированию фермента. Ниже показаны структуры стабильных ацилферментов, получающихся при ингибировании сериновых протеиназ фенилметилсульфонилфторидом (слева) и диизопропилфторфосфатом (справа):

Изложенные выше представления о механизме действия сериновых протеиназ подтверждены методами белковой инженерии. Замена серина активного центра в ряде ферментов этого семейства (при помощи сайт-специфичного мутагенеза) на аланин неизменно приводит к практически полной утрате активности. Замена компонентов каталитического центра в субтилизине (протеиназе бацилл) дала следующие результаты. Замена серина Ser-221 (остатка, соответствующего Ser-195 в химотрипсине) аланином снижала активность фермента примерно в 106 раз, причем практически весь эффект приходился на снижение каталитической константы kкат, а константа Михаэлиса сохраняла свое значение. Такие же результаты давала замена гистидина каталитического центра аланином. Несколько меньшим, но все же очень значительным было падение kкат при замене Asp-32 аланином, kкат в этом случае составляла 4∙10-6 от величины, характеризующей исходный фермент.
Таким образом, все компоненты «триады» каталитического центра критически важны для эффективного катализа. Роль остатка аспарагиновой кислоты, которая может показаться менее значимой с учетом приведенного выше механизма, видимо, состоит в том, что карбоксилат-анион этого остатка, во-первых, фиксирует в пространстве имидазольное кольцо гистидина каталитического центра и, во-вторых, обеспечивает закрепление приведенного выше на схемах таугомерного состояния имидазольной группы.
Труднее анализировать этим методом роль N—Н-групп, образующих оксианионную впадину, так как в химотрипсине и гомологичных ему ферментах обе они принадлежат пептидному скелету. Однако в субтилизине роль одного из компонентов оксианионной впадины выполняет амидная группа аспарагина. Замена этого остатка лейцином снижает эффективность поляризующего действия оксианионной впадины на С = О-группировку субстрата, вследствие чего активность снижается примерно в 103 раз, сохраняя тем не менее вполне заметный уровень