Основы биохимии - А. А. Анисимов 1986
Липиды
Терпены
8.7.1. Общая характеристика терпенов. В основе терпеновых соединений лежат изопреновые остатки, чаще всего соединенные между собой «голова к хвосту», но иногда возможно апериодическое расположение изопреновых единиц — «хвост к хвосту».

К терпенам относятся эфирные масла (лимонен, пинен, гераниол, камфора, ментол), смоляные кислоты и каучук, различные растительные пигменты (каротины, ликопин и др.), а также витамин А и сквален из животных тканей. Химически родственны терпенам витамин Е, витамин К, кофермент Q (см. разд. 7.3.1).
В зависимости от числа изопреновых группировок все терпены делятся на монотерпены (2 группировки), сесквитерпены (3), дитерпены (4), тритерпены (6) и тетратерпены (8). Молекулы терпенов могут обладать линейной или циклической структурой, у некоторых из них встречаются как те, так и другие компоненты. Возможны взаимные переходы из одной структуры в другую внутри каждой группы терпенов. Двойные связи в линейных сегментах находятся у большинства терпенов в устойчивой транс-конфигурации, но возможна и цис-конфигурация одной или нескольких двойных связей. Например, в печени обнаружена цис-форма витамина А, а в зрительном пигменте глаза — цис-форма каротиноида.
Физиологическая роль терпенов выяснена недостаточно. Однако из того, что известно, следует, что функции их разнообразны. Так, эфирные масла, смоляные кислоты, гутта, каучук выполняют защитную функцию; пигменты принимают участие в одном из важнейших процессов растительного организма — фотосинтезе, а также придают окраску цветам и плодам; некоторые из терпенов являются витаминами. Сквален — тритерпен — главный компонент секрета сальных желез, основной липидный компонент вируса дифтерии птиц, жира акульей печени, а также промежуточный продукт в биосинтезе холестерина.
Некоторые монотерпены у членистоногих являются феромонами, которые представляют собой средство информации между насекомыми, в частности являются половыми аттрактантами. Основные компоненты феромонов — ненасыщенные углеводороды, а также высокомолекулярные спирты и их эфиры. Феромоны имеют огромное значение не только в плане теоретического изучения поведения насекомых, но и практического использования в сельском хозяйстве, медицине, лесном хозяйстве для борьбы с вредными насекомыми.
8.7.2. Эфирные масла и смоляные кислоты. Среди эфирных масел большое распространение имеют монотерпены С10Н16. Они могут быть как алифатического ряда, так и циклического. В природе больше распространены кислородные производные алифатических терпенов — альдегиды и спирты. Эфирным маслам присущ резкий и обычно приятный запах, летучесть (способность перегоняться с водяным паром), высокий коэффициент оптического преломления (1,4—1,6).
Среди кислородных производных терпенов можно назвать лина-лоол — жидкость с запахом ландыша:

Обнаружен в кожице цитрусовых, в цветах ландыша. Применяется в парфюмерии сам и его эфиры.
Близки по строению к предыдущему гераниол и цитронеллол. Последний обладает запахом розы, в большом количестве содержится в розовом и гераниевом маслах. 80% всей мировой продукции розового масла добывают из одного вида Rosa damascena. Для получения 1 кг розового масла расходуется 35 млн. лепестков роз.
Из циклических монотерпенов большой интерес представляет ментол. На его долю приходится 70% в составе масла мяты. Ментол обладает свойством раздражать нервные окончания; оказывает легкое местное обезболивание и слабое антисептическое действие. Его применяют в составе сердечных средств (валидол, капли Зеленина), при насморке и других заболеваниях верхних дыхательных путей, при мигрени (ментоловый карандаш).
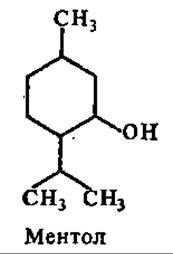
Среди бициклических монотерпенов важную роль играет пинен — главный компонент скипидара, получаемого из сосновой живицы. Широко применяется в качестве растворителя в лакокрасочной промышленности и в живописи. К этой же группе терпенов относится камфора. Очень высока концентрация камфоры в древесине и листьях камфарного лавра, в некоторых видах полыни, откуда ее и получают. Для синтетического получения используют скипидар. Применяют в медицине для возбуждения центральной нервной системы, стимуляции дыхания и кровообращения. Камфора оказывает раздражающее и антисептическое действие, в связи с чем употребляется в виде мазей и втираний при воспалительных процессах и ревматизме. Используется как сырье в ряде отраслей химической промышленности.

Смоляные кислоты относятся к дитерпенам. Имеют формулу С20Н30О2 и составляют 4/5 смолы хвойных растений — терпентина (живица). При переработке живицы с водяным паром отгоняется скипидар, и остается твердый остаток — канифоль, основную массу которой составляют «смоляные кислоты», в частности абиетиновая кислота
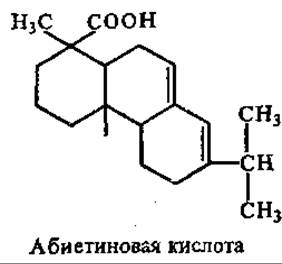
Смоляные кислоты по некоторым свойствам аналогичны жирным кислотам. Их Na- и К-соли обладают мылящими свойствами, поэтому из канифоли иногда приготовляют мыла с высокой пенообразующей способностью.
Канифоль служит сырьем для многих отраслей химической промышленности (производство резин, пластмасс, искусственной кожи, линолеума, сургуча, лаков, красок, лыжной мази). Из нее получают электроизоляционные мастики и компаунды. Она служит флюсом при лужении и пайке металлов. Музыканты, играющие на смычковых инструментах, натирают ею смычок.
В состав выделяемых растениями смол кроме смоляных кислот входят также смоляные спирты, фенолы, углеводороды.
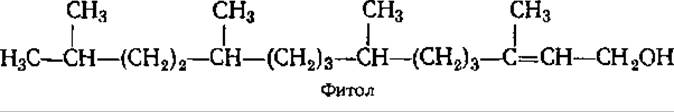
К дитерпенам относятся также фитол, витамин А, гиббереллины (см. разд. 10.2 и 12.7). Фитол в свободном виде в природе не встречается, а входит в качестве терпеновой части в состав хлорофилла, витаминов К и Е:
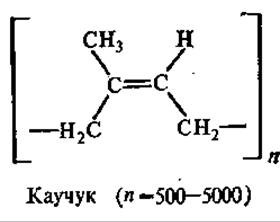
Каучук и еутта — политерпены. Главным источником каучука служит тропическое каучуконосное дерево гевея, а основные отечественные каучуконосы — кок-сагыз и тау-сагыз из семейства сложноцветных. Гуттаперченосом являются некоторые виды бересклета и эвкомия.
Каучук и гутта состоят из остатков изопрена, связанных «голова к хвосту». Полиизопреновая цепочка каучука имеет цис-конфигурацию, а цепочка гуттаперчи (его изомер) — транс-конфигурацию. Различия в строении каучука и гуттаперчи обусловливают и отличия в их физических свойствах. Так, каучук при обычной температуре эластичен и аморфен, а при охлаждении приобретает кристаллическую структуру. При комнатной температуре гуттаперча — твердый кожеподобный продукт, а при температуре 50—100°С становится пластичной.
Каучук и гуттаперча находят широкое применение в резиновой промышленности, радио- и электропромышленности в качестве изоляционного материала, а также при изготовлении кислотоупорных и клеящих материалов.
8.8. Обмен липидов
8.8.1. Превращения липидов в процессе пищеварения и всасывание. Липиды — важная составная часть пищи. Взрослому человеку требуется от 70 до 145 г жира в сутки в зависимости от трудовой деятельности, пола, климатических условий. Причем необходимы как животные, так и растительные жиры. Липиды являются высокоэнергетическими веществами, поэтому за их счет удовлетворяется 25—30% потребности человеческого организма в энергетическом материале. Кроме того, в составе животных жиров в организм поступают жирорастворимые витамины A, D, К и Е, растительные жиры богаты непредельными жирными кислотами (витамин F), являющимися предшественниками простагландинов, исходным материалом для синтеза организмом фосфолипидов и других веществ.
Переваривание жира начинается в желудке, где находится фермент липаза. Но она расщепляет только эмульгированный жир, каким является жир молока, и поэтому этот процесс имеет значение главным образом у детей грудного возраста.
Существуют данные, что у молодняка жвачных животных и крыс липазная активность обнаруживается в слюне. Вполне возможно, что в некоторых случаях липолитическая активность желудка может быть обусловлена липазой, секретируемой в ротовой полости и попадающей в желудок вместе со слюной.
Основное расщепление липидов происходит в кишечнике, в первую очередь в двенадцатиперстной кишке. В этот отдел кишечника поступает сок поджелудочной железы, содержащий очень активную липазу. Сюда же поступает из желчного пузыря желчь, составные компоненты которой (желчные кислоты) необходимы для переваривания липидов. Это связано с тем, что желчные кислоты — холевая (преобладает в желчи человека), дезоксихолевая, литохолевая, хенодезоксихолевая, таурохолевая и гликохолевая — представляют собой поверхностно-активные вещества, способствующие эмульгированию жиров, что является важнейшим условием их последующего ферментативного расщепления (рис. 8.1).
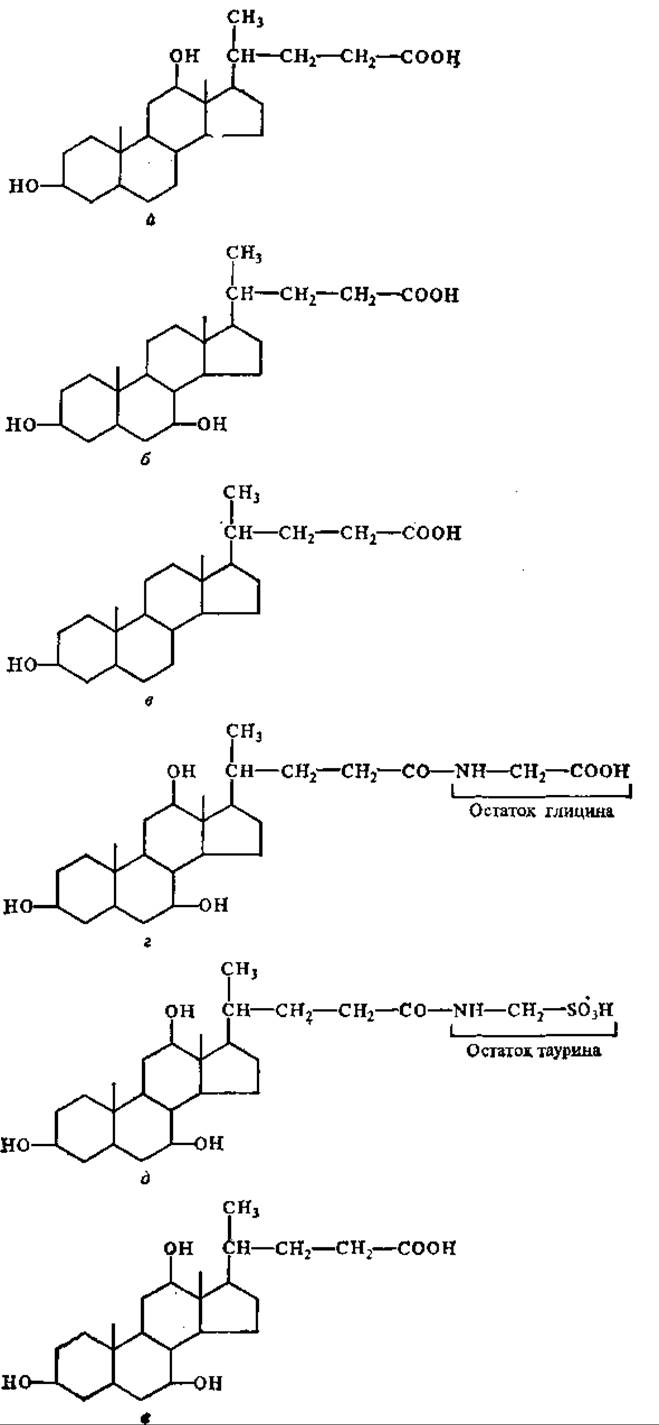
Рис. 8.1. Желчные кислоты:
а — дезоксихолевая, б — хенодезоксихолевая, в — литохолевая, г — гликохолевая, д — таурохолевая, е — холевая
Пройдя через барьер слизистой оболочки кишечника, желчные кислоты в связанном состоянии с липидами отделяются от последних и по венам кишечника через портальный кровоток возвращаются в печень, а затем с желчью в двенадцатиперстную кишку.
Образование эмульсии жиров в кишечнике может происходить и под влиянием мелких пузырьков СО2, выделяющегося при нейтрализации соляной кислоты пищевой кашицы бикарбонатами поджелудочного и кишечного сока. Способствуют эмульгированию и соли жирных кислот (мыла), возникающие при гидролизе липидов. Но основная роль в эмульгировании жиров принадлежит желчным кислотам.
В результате описанных процессов образуется очень тонкая жировая эмульсия, диаметр частиц которой не превышает 0,5 мкм. Такие эмульгированные жиры способны самостоятельно проходить через стенку кишечника и попадать в лимфатическую систему. Однако большая часть эмульгированного жира всасывается после гидролитического расщепления его панкреатическими липазами. Последние образуются в поджелудочной железе в виде неактивных проферментов, которые переходят в активную форму при участии желчных кислот.
Основная масса липидов пищи представлена триацилглицеринами, меньше фосфолипидами и стероидами. Гидролиз триацилглицеринов идет постепенно. Сначала расщепляются эфирные связи в 1-м и 3-м положениях, т. е. внешние сложноэфирные связи;
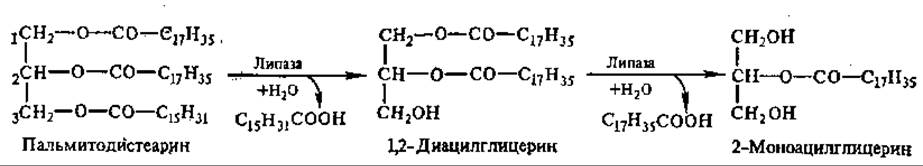
Эти реакции осуществляют липазы, специфичные в отношении 1,3-эфирных связей триацилглицерина. Связи во 2-м положении гидролизуют другие липазы:
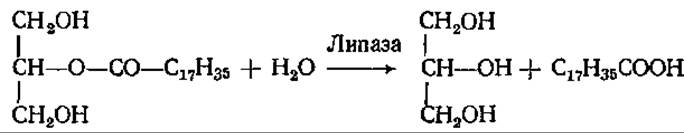
Связи 1 и 3 гидролизуются быстро, а потом идет медленный гидролиз 2-моноглицерида. 2-Моноглицерид может всасываться стенкой кишечника и использоваться на ресинтез триацилглицеринов, специфичных для данного вида организмов, уже в самой слизистой тонкого кишечника. Таким образом, ресинтез жиров происходит в процессе их всасывания через стенку кишечника.
Кроме липаз в соке поджелудочной железы присутствуют эстеразы, гидролизующие преимущественно эфиры жирных кислот с короткой цепью (например, трибутирин) и эфиры холестерина. Эти эстеразы тоже активны только в присутствии желчных кислот.
Пищеварительные липазы кроме человека и млекопитающих животных обнаружены и исследованы у рыб, некоторых беспозвоночных. Однако, как правило, у большинства видов беспозвоночных и костистых рыб липолитическая активность в пищеварительных соках примерно в 1000 раз ниже, чем в панкреатическом соке млекопитающих. Не следует забывать, что жиры могут усваиваться также путем фагоцитоза и сохраняться без предварительного гидролиза до тех пор, пока не прогидролизуются внутриклеточными липазами и, таким образом, примут участие в синтезе липидов в процессах образования энергии.
Интересным является вопрос, на каком этапе эволюции внутриклеточное расщепление жиров перешло во внеклеточное и как различаются липазы этих процессов. У животных, для которых характерно пищеварение смешанного типа, например у многих двустворчатых моллюсков, вне- и внутриклеточные липазы очень сходны по свойствам. Можно предполагать, что липазы поджелудочной железы произошли из внутриклеточных ферментов и специализировались в конкуренции за овладение просветом кишечника с помощью более сильного сродства к поверхности раздела масло — вода и более развитой полярной структуры молекулы фермента, что обеспечивает необходимую ориентацию на поверхности раздела. Необходимость в большей активности или более высокой концентрации внеклеточных липаз высших животных вызвана, видимо, тем, что у них должно усваиваться и расщепляться большее количество жиров в течение более короткого времени.
Расщепление фосфолипидов происходит при участии ряда ферментов: фосфолипаз А1, А2, С, D и лизофосфолипазы.
Фосфолипаза а1 гидролизует связь в 1-м положении. Фосфолипаза A2, образующаяся в поджелудочной железе в виде профермента, который затем активируется трипсином, отщепляет жирную кислоту во 2-м положении. В результате ее действия образуются лизофосфолипиды (см. разд. 8.8.3), которые являются сильными поверхностно-активными веществами и обладают мощным гемолитическим действием (лизофосфатидилхолин, лизофосфатидилэтаноламин). Кроме панкреатического сока фосфолипаза А2 содержится в ядах рептилий, беспозвоночных (особенно членистоногих — пчел, скорпионов, муравьев), а также у кишечнополостных. Известны также внутриклеточные фосфолипазы А2 (в лизосомах, микросомах, митохондриях).
Лизофосфолипаза катализирует гидролиз одной сложноэфирной связи в положении 1, превращая лизофосфолипиды в соответствующие глицерофосфорильные производные. Фосфолипаза С вызывает гидролиз связи между фосфорной кислотой и глицерином, а фосфолипаза D отщепляет полярную Х-группу. Фосфолипаза С пищеварительного тракта животных недостаточно изучена, пока обнаружена в небольшом числе объектов (в слизистой кишечника морской свинки, в поджелудочной железе быка). Широко распространены и хорошо исследованы экзогенные бактериальные фосфолипазы С.
Стериды, подвергаясь действию гидролитических ферментов типа холестераз, расщепляются в кишечнике с образованием спирта холестерола или эргостерола и соответствующей жирной кислоты. Холестеразы продуцируются поджелудочной железой и активны только в присутствии солей желчных кислот.
Таким образом, образующаяся в результате гидролиза липидов смесь содержит анионы жирных кислот, моно-, ди- и триацилглицерины, хорошо эмульгированные солями жирных кислот и мылами, глицерин, холин, этаноламин и другие полярные компоненты липидов. Исследования с мечеными триацилглицеринами показали, что около 40% жиров пищи гидролизуется полностью до глицерина и жирных кислот, 3—10% всасываются без гидролиза в форме триацилглицеринов, а остальные гидролизуются частично, главным образом до 2-моноацилглицеринов. Глицерин водорастворим и вместе с жирными кислотами, имеющими короткие углеродные цепи (С < 10), уходит из кишечника через портальную систему кровообращения и поступает в печень. Такие жирные кислоты всасываются преимущественно в неэтерифицированной форме.
Для всасывания жирных кислот с длинной цепью (С > 10) необходимы желчные кислоты. Способность последних транспортировать жирные кислоты через слизистую оболочку кишечника связана с их поверхностно-активными свойствами и способностью к мицеллообразованию с моноацилглицеринами и мылами. Такие мицеллы могут проходить через водный слой, имеющийся на поверхности слизистой оболочки кишечника. Особую роль при этом играют такие желчные кислоты, как таурохолевая и гликохолевая. Соединяясь с жирными кислотами, они образуют растворимые комплексы — холеиновые кислоты, которые легко всасываются в эпителий кишечника. Так как pH в тонком кишечнике слабощелочная, желчные кислоты функционируют здесь в форме своих солей.
Лучше перевариваются и всасываются липиды, находящиеся в жидком состоянии, при температуре тела. Липиды, у которых точка плавления существенно выше температуры тела, плохо перевариваются и всасываются.
Фосфорная кислота, образующаяся при гидролизе фосфолипидов, всасывается в виде солей, а азотистые основания — холин, этаноламин и серин — всасываются при участии нуклеотидов (ЦДФ).
Некоторая избирательность проявляется слизистой оболочкой кишечника в отношении стероидов, особенно растительного происхождения. Среди основных стероидов пищи только холестерин легко проникает через стенки кишечника. С такой же легкостью всасываются витамин D и некоторые стероидные гормоны, введенные перорально.
Преобладающими липидами лимфы являются триацилглицерины, даже тогда, когда жирные кислоты находятся в составе сложных эфиров других спиртов.
Ресинтезированные триацилглицерины, фосфолипиды, холестерин и его эфиры в эпителиальных клетках кишечника соединяются с небольшим количеством белка и образуют хиломикроны (ХМ). Благодаря большим размерам (100—5000 нм) ХМ не способны проникать в кровеносные капилляры и диффундируют в лимфатическую систему кишечника, а из нее через грудной лимфатический проток в кровяное русло, т. е. с их помощью осуществляется транспорт экзогенных триадилглицеринов, холестерина и частично фосфолипидов в печень и жировую ткань. Хиломикроны свободно диффундируют из плазмы крови в межклеточные пространства печени. Гидролиз триадилглицеринов может происходить как внутри клеток печени, так и на их поверхности. В клетки жировой ткани ХМ не способны проникать, в связи с чем триадилглицерины гидролизуются на поверхности эндотелия капилляров жировой ткани при участии фермента липопротеинлипазы. Часть образовавшихся жирных кислот проходит внутрь клеток и запасается в виде соответствующих липидов, другая часть связывается с альбуминами сыворотки крови и разносится в другие органы и ткани. С током крови может покидать жировую ткань и глицерин.
8.8.2. Окисление глицерина и жирных кислот в тканях. Реакции окисления. Резервные липиды, состоящие более чем на 99% из триадилглицеринов, непрерывно мобилизуются, но одновременно такое же количество вновь депонируется.
Метаболизм резервных липидов начинается с гидролиза под действием липопротеинлипазы плазмы крови или активируемой гормонами липопротеинлипазы жировой ткани. Образующиеся жирные кислоты поступают в кровоток и транспортируются в связанном с сывороточными альбуминами состоянии. Вход свободных жирных кислот сопровождается появлением в плазме также и глицерина, но в значительно меньшем количестве, чем соответствующих жирных кислот. Глицерин может участвовать в глюконеогенезе или включаться в гликолитический путь расщепления с предварительным образованием глицерол-3-фосфата под действием глицеролкиназы и при участии АТФ:

Главное место деградации жирных кислот до ацетил-КоА — печень. После того как жирные кислоты поступают в клетку, они должны быть активированы путем образования коэнзим А-производных. Реакция активации может осуществляться двумя способами.
В первом случае, характерном для животных тканей, ацил-КоА — синтетаза катализирует образование КоА-производных жирных кислот согласно реакции
![]()
Известно несколько таких ферментов. Они именуются в соответствии с названием жирной кислоты, скорость превращения которой данным ферментом максимальна. Например, ацетил-КоА — синтетаза катализирует превращение С2 и С3 жирных кислот. Ацил-КоА — синтетазы присутствуют в эндоплазматической сети и в наружной митохондриальной мембране. В митохондриальном матриксе содержится ацил-КоА — синтетаза, активирующая жирные кислоты с длинной цепью и использующая ГТФ вместо АТФ. Продуктами реакции в этом случае являются ацил-КоА, ГДФ и неорганический фосфат.
В ряде растений была также обнаружена ацетил-КоА — синтетаза, имеющая общие свойства с аналогичным ферментом из животных тканей. В то же время у некоторых микроорганизмов преобладает другой способ активации жирных кислот с короткой цепью:
![]()
![]()
Здесь активация осуществляется с помощью КоА-трансферазных реакций.
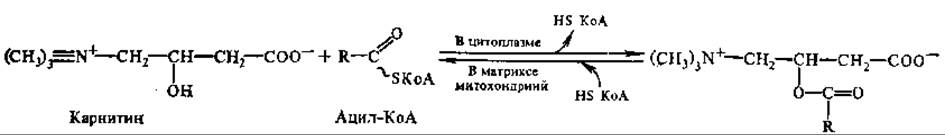
Существуют и некоторые другие пути образования ацил-производных КоА. Окисление этих производных происходит только в митохондриях, однако проникать в митохондрии они самостоятельно не способны. Важную роль в транспорте КоА-производных жирных кислот из цитоплазмы в матрикс митохондрий играет карнатин (витамин Вт, L-3-гидрокси-4-триметиламмонийбутират):
![]()
ь этом процессе участвуют две ацилтрансферазы: карнитин-ацетилтрансфераза (ацилирует карнитин жирными кислотами с короткой цепью) и карнитин-пальмитоилтрансфераза (ацилирует карнитин кислотами с длинной цепью). Оба фермента локализуются как в цитоплазме, так и на наружной и внутренней мембранах митохондрий и осуществляют перенос остатков жирной кислоты от ацил-КоА на карнитин. Ацилкарнитин легко проникает через мембрану и внутри митохондрии передает жирную кислоту на внутримитохондриальный КоА, при этом высвобождается карнитин. Реакция катализируется внутримитохондриальной карнитин-ацилтрансферазой. В результате достигается разделение цитоплазматического и митохондриального пулов КоА.
КоА-производные жирных кислот в матриксе подвергаются окислению путем ряда последовательно повторяющихся реакций, которые в каждом повторении приводят к укорачиванию цепи жирной кислоты на два углеродных атома (ß-окисление)1. Все промежуточные продукты при ß-окислении являются производнымиКоА. Этот тип окисления жирных кислот — основной путь распада жирных кислот как в животных, так и в растительных тканях.
1 По Международной номенклатуре положение замещающих групп в карбоновых кислотах отмечается не буквами греческого алфавита, как ранее, а цифрами. Однако старое название процесса окисления жирных кислот — ß-окисление пока сохранилось.
Следующей реакцией на пути расщепления жирных кислот является дегидрирование, катализируемое различными ФАД-содержащими ацил-КоА-дегидрогеназами с образованием транс-2,3-ненасыщенных производных:

Для нормальной работы ФАД-зависимых дегидрогеназ необходим переход атомов водорода от их флавиновой части к электронпереносящему флавопротеину, который в свою очередь направляет электроны в электронтранспортную цепь.
Далее происходит гидратация двойной связи по реакции

На следующем этапе цикла следует второе дегидрирование КоА-эфира L-3-гидроксикислоты с образованием 3-кетопроизводных:
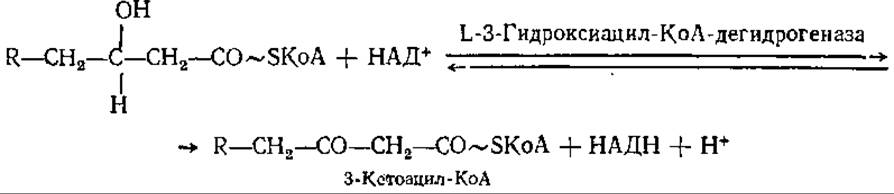
Фермент, катализирующий эту реакцию, абсолютно специфичен к L-стереоизомеру КоА-эфиров 3-гидроксикислот, но не специфичен в отношении числа атомов углерода в молекуле жирной кислоты.
И, наконец, в последней стадии каждого из повторяющихся циклов окисления жирных кислот 3-кето-ацил-КоА вступает в реакцию с другой молекулой КоА, в результате чего образуются молекулы ацетил-КоА и жирнокислотного производного КоА, укороченного на два углеродных атома:
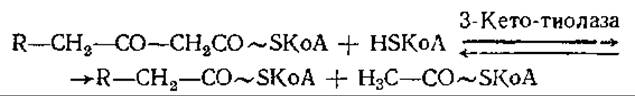
Эта реакция носит название тиолиза и является сильно экзергонической, поэтому равновесие в ней всегда смещено в правую сторону. Некоторые из известных тиолаз проявляют специфичность в отношении длины цепи.
Последовательное повторение этого ряда реакций приводит н полному распаду жирных кислот с четным числом атомов углерода до ацетил-КоА. Жирные кислоты с нечетным числом атомов углерода распадаются до соответствующего числа молекул ацетил- КоА и одной молекулы пропионил-КоА.
Окисление ненасыщенных жирных кислот происходит так же, как и насыщенных, но после удаления нескольких единиц ацетил- КоА образуется ∆3,4-ацил-КоА, а не ∆2,3-ацил-КоА и, чтобы окисление прошло полностью, необходим еще один фермент — ∆3,4-цис-∆2,3-транс-еноил-КоА-изомераза, который катализирует перемещение двойной связи из положения 3—4 в положение 2—3 и изменение конфигурации этой связи из цис- в транс-. В результате образуется нормальный субстрат, который может в дальнейшем окисляться путем последовательного удаления ацетил-КоА. Так, например, окисляется олеиновая кислота.
Полиненасыщенные жирные кислоты требуют для своего полного окисления кроме ∆3,4-цис-∆2,3-транс-еноил-КоА-изомеразы еще один дополнительный фермент — 3-гидроксибутирил-КоА-эпимеразу, которая катализирует превращение D-стереоизомера 3- гидроксиацил-КоА в L-стереоизомер, легко окисляемый L-специфичной 3-гидроксиацил-КоА-дегидрогеназой. D-Форма 3-гидроксиацил-КоА возникает при гидратации образующегося в ходе последовательного отщепления ацетил-КоА от полиненасыщенной жирной кислоты ∆2,3-цис-ацил-КоА под действием фермента 3-гидрокси-КоА-гидро-лиазы.
Таким образом, наличие изомеразы и эпимеразы обеспечивает возможность полного окисления всех ненасыщенных жирных кислот, содержащихся в природных липидах.
Баланс энергии. При окислении жирных кислот до конечных продуктов выделяется большое количество энергии. Например, пальмитиновая кислота: при окислительном расщеплении одной молекулы пальмитоил-КоА до восьми молекул ацетил-КоА необходимо 7 следующих друг за другом циклов, в каждом из которых образуется одна молекула восстановленного ФАД, отдающего в дыхательную цепь пару электронов на уровне KoQ или цитохрома b, в результате чего синтезируется две молекулы АТФ. Образующаяся в каждом цикле одна молекула НАДН + Н+ приводит к синтезу еще трех молекул АТФ.
Суммарное уравнение имеет следующий вид:
Пальмитоил-КоА + 7КоА + 7ФАД + 7НАД+ + 7Н2O →
→ 8 Ацетил-КоА + 7ФАДН2 + 7НАДН + 7Н+
При сопряжении окисления с фосфорилированием это уравнение будет выглядеть так:
Пальмитоил-КоА + 7КоА + 7O2 + 35ФН + 35АДФ →
→ 8 Ацетил-КоА + 35 АТФ + 42 Н2O
Однако образовавшиеся в результате окисления пальмитиновой кислоты восемь молекул ацетил-КоА вступают в цикл трикарбоновых кислот и баланс, включающий окисление ацетил-КоА и сопряженное с ним фосфорилирование, выражается следующим уравнением:
8 Ацетил-КоА + 16 О2 + 96ФН + 96АДФ → 8КоА +
+ 96АТФ + 104Н2О + 16СO2
В результате суммирования двух последних уравнений получается уравнение
Пальмитоил-КоА + 23O2 + 131 ФH + 131 АДФ →
→ КоА + 16СO2 + 146 Н2O + 131 АТФ
Если учесть, что в самом начале на активацию пальмитиновой кислоты тратится одна молекула АТФ (или ГТФ), то суммарный выход на одну молекулу пальмитиновой кислоты составляет 130 молекул АТФ.
Если принять стандартное ∆G° для гидролиза АТФ равным примерно 34,5 кДж, a ∆G° при полном окислении пальмитата до Н2O и СО2 равно 9797 кДж, то коэффициент полезного действия биологического окисления пальмитата составит 34,5х130x 100%/9797 = 45%. Таким образом, эффективность накопления энергии как при окислении пальмитиновой кислоты, так и глюкозы примерно одинакова, но при расчете на единицу массы при окислении пальмитата АТФ образуется в два раза больше (0,51 моль АТФ/г), чем при окислении глюкозы (0,21 моль АТФ/г).
а- и ω-Oкисление жирных кислот. Наряду с ß-окислением высокомолекулярные жирные кислоты (от C13 ДО C18) могут подвергаться а-окислению. Этот тип окисления особенно характерен для растительных тканей, но может происходить и у животных, в частности в микросомах мозга и других нервных тканей, в состав которых в большом количестве входят цереброзиды и ганглиозиды, содержащие жирные 3-гидроксикислоты.
Суть а-окисления заключается в том, что происходит гидроксилирование жирной кислоты во 2-м положении под действием митохондриальной монооксигеназы (необходим О2, Mg2+, НАДФН и кофактор). Гидроксикислота далее превращается в 2-кетокислоту, которая подвергается окислительному декарбоксилированию уже в эндоплазматической сети с образованием СO2 и жирной кислоты с числом атомов углерода на один меньше, чем в исходной.
R—СН2—СН2—СН2—СООН → R—CH2—СН2—СН(ОН)—COOH→
→R—CH2—CH2—CO—СООН → R—CH2—CH2—COOH + CO2
Образующаяся кислота может затем включаться в аналогичную реакцию, но полного окисления жирных кислот до СО2 и Н2O по a-механизму не происходит из-за специфичности фермента, способного использовать в качестве субстрата только молекулы с длиной цепи от 13 до 18 углеродных атомов. а-Окисление энергетически не столь выгодно, как ß-окисление.
Некоторые жирные кислоты менее длинноцепочечные или со средней длиной цепи могут подвергаться окислению и по ω-углеродному атому с образованием в качестве конечного продукта а, (ω-дикарбоновых кислот. Этот путь называется ω-окислением. Образующаяся дикарбоновая кислота может быть укорочена с любого конца молекулы с помощью ß-окисления, описанного выше.
У некоторых микроорганизмов образование а, ω-дикарбоновых кислот может происходить путем расщепления двойной связи. Именно таким путем из олеиновой кислоты образуется азелаиновая кислота (С9), которая подвергается ß-окислению, давая пимелиновую кислоту — предшественник биотина.
Метаболизм пропионовой кислоты. При окислении одной молекулы жирных кислот с нечетным числом атомов углерода образуется кроме ацетил-КоА еще одна молекула пропионил-КоА. Распад жирных кислот и аминокислот (валин, изолейцин) с разветвленной цепью также ведет к образованию пропионовой кислоты или пропионил-КоА.
Известно несколько путей метаболизации пропионата. Один из них характерен для животных тканей и заключается в карбоксилировании пропионил-КоА с образованием метилмалонил-КоА, который с участием специфической мутазы превращается в сукцинил-КоА. Дальнейшие превращения образующегося сукцинил-КоА связаны с циклом трикарбоновых кислот.
Второй путь окисления пропионовой кислоты обнаружен как в растениях, так и у животных и бактерий, он называется видоизмененным ß-окислением. Его начальные стадии вплоть до образования 3-гидроксипропионил-КоА те же самые, что и при обычном ß-окислении. В дальнейшем в результате реакций окисления и окислительного декарбоксилирования образуется ацетил-КоА.
Кетоновые тела. Обычно деградация жирных кислот происходит без существенного накопления промежуточных продуктов и, в частности, ацетил-КоА. Однако некоторые условия (голодание, сахарный диабет, резкие смены диеты и т. п.) приводят к появлению в крови так называемых кетоновых тел. К ним относятся ацетоуксусная кислота, 3-гидроксимасляная кислота и ацетон. Образуются они в печени из ацетил-КоА и током крови доставляются к периферическим тканям, где окисляются в цикле трикарбоновых кислот. Потеря организмом способности утилизировать эти вещества (кетоз) приводит к их значительному накоплению в крови (кетонемия) и в моче (кетонурия), что является диагностическим признаком ряда заболеваний.
Ацетоуксусная кислота появляется в результате конденсации двух молекул ацетил-КоА по принципу «голова к хвосту». Реакция катализируется ферментом ацетил-КоА — ацетилтрансферазой.
Ацетил-S-KoA + Ацетил-S-KoA Ацетоацетил-S-KoA + HS-KoA
Образующийся ацетоацетил-КоА может превращаться в свободную ацетоуксусную кислоту двумя путями. Он может взаимодействовать еще с одной молекулой ацетил-КоА при участии фермента гидроксиметилглутарил-КоА — синтазы с образованием 3-гидрокси-3-метилглутарил-КоА, который под действием гидроксиметилглутарил-КоА — лиазы расщепляется на ацетоацетат и ацетил-КоА. Кроме того, ацетоацетил-КоА способен превращаться в свободную ацетоуксусную кислоту, отщепляя HS-KoA путем деацилирования. Реакция катализируется ацетоацетил-КоА — гидролазой.
При восстановлении ацетоацетата образуется D-3-гидроксимасляная кислота:
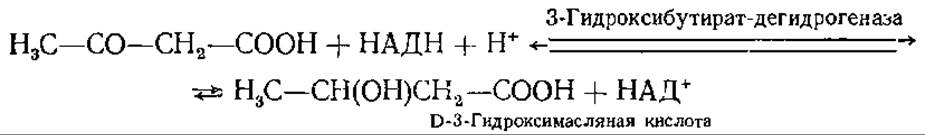
Ацетон образуется из ацетоацетата при декарбоксилировании, катализируемом ацетоацетатдекарбоксилазой:
![]()
Соотношение кетоновых тел в кровотоке колеблется: если в печени много гликогена, то предпочтительно образуется 3-гидроксимасляная кислота, когда его мало, преобладает ацетоацетат.
Метаболизация восков, их роль в пищевых цепях. За небольшим исключением все животные, как позвоночные, так и беспозвоночные, не способны переваривать пчелиный воск. Переваривание происходит с участием симбиотических бактерий, например у гусеницы паразита пчелиных ульев — большой восковой огневки.
В целом, воска занимают очень важное место в пищевых цепях морских животных. Первичными производителями их являются, видимо, мелкие планктонные ракообразные, особенно веслоногие. У некоторых из них воска составляют до 70% сухой массы тела, хотя питаются они фитопланктоном, не содержащим воска. Поскольку планктонные ракообразные являются главным связующим звеном между микроскопическим фитопланктоном и крупными обитателями моря, считают, что примерно половина всего органического вещества, образующегося на Земле в процессе фотосинтеза, на какое-то время превращается в воска. Рыбы, питающиеся веслоногими (сельди, сардины), имеют в пищеварительном тракте липазы для расщепления восков. Спирты последних окисляются до жирных кислот, которые затем используются на биосинтез нейтральных жиров.
8.8.3. Биосинтез липидов. У большинства организмов биосинтез липидов — важное звено обмена веществ. В виде жирных кислот и триацилглицеринов в организме животных запасается в больших количествах энергетический материал, так как «запасающая емкость» для углеводов в организме невелика. В семенах растений также накапливается много триацилглицеринов, которые при прорастании используются как энергетический, так и пластический материал. Важным моментом в синтезе липидов является образование полярных липидов, входящих в состав мембран, поскольку в большинстве клеток они постоянно обновляются.
Биосинтез жирных кислот. Синтез насыщенных и мононенасыщенных жирных кислот легко происходит в любом живом организме. Основным местом их синтеза в отличие от окисления, локализованного только в митохондриях, является цитоплазма. В некотором количестве жирные кислоты синтезируются также с участием ферментов, локализованных в мембранах эндоплазматической сети путем удлинения КоА-производных полиненасыщенных жирных кислот. Известен и еще один тип синтеза, при котором происходит удлинение молекул жирных кислот в митохондриях. Особенно активно липогенез протекает в жировой ткани, молочной железе, печени. Сырьем для биосинтеза служит ацетил-КоА, который образуется из избыточной глюкозы пищи, не использованной на энергетические нужды, сохраняемых в резерве полисахаридов и аминокислот, не требующихся для других функций. В качестве восстановителя в биосинтезе жирных кислот расходуется НАДФН, образующийся в пентозофосфатном цикле (см. разд. 6.9.3). Другим источником НАДФН служит окисление малата до пирувата и СО2 малатдегидрогеназой. В листьях растений поставщиком НАДФН, необходимого для синтеза жирных кислот, является процесс фотовосстановления НАДФ+.
Основной путь биосинтеза жирных кислот, протекающий в цитоплазме, катализируется особой синтетазной системой, состоящей из 7 ферментов. Конечный продукт этого пути — пальмитиновая кислота, представляющая собой источник всех других насыщенных и мононенасыщенных жирных кислот млекопитающих и всех жирных кислот микроорганизмов.
Суммарная реакция биосинтеза жирных кислот в цитоплазме имеет следующий вид:

Как видно из реакции, для синтеза требуется единственная молекула ацетил-КоА, которая служит затравкой. Активную роль в биосинтезе жирных кислот играет особый ацилпереносящий белок (АПБ). У Е. coli он обладает небольшой молекулярной массой, относится к сложным белкам, простетической группой у него является 4-фосфопантотеновая кислота и тиоэтиламин с его сульфгидрильной группой. Пантотеновая часть молекулы АПБ действует как «рука», переносящая растущую цепь жирной кислоты от одного фермента к другому. Полагают, что у млекопитающих активность АПБ присуща определенным участкам отдельных полипептидных цепей, которые обладают также и другими активностями, требующимися для биосинтеза жирных кислот.
Процесс начинается с переноса ацетильной группы ацетил-КоА на 4-фосфопантотеин-АПБ (к его тиоловой группе). С карбоксильной группы этого первого ацетила начинается рост цепи жирной кислоты путем последовательного добавления ацетильных остатков. Другие семь ацетильных групп добавляются из малонил-КоА, образующегося с помощью ацетил-КоА — карбоксилазы из ацетил-КоА и бикарбоната:
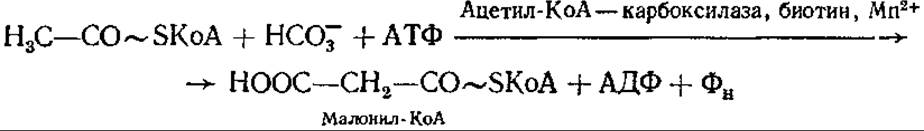
Ацетил-КоА — карбоксилаза содержит в качестве простетической группы витамин биотин. Реакция образования малонил-КоА протекает в две стадии: сначала карбоксилируется биотиновая часть карбоксилазы, а затем фермент транскарбоксилаза переносит СО2 с биотина на ацетил-КоА, образуя малонил-КоА. У некоторых микроорганизмов малонил-КоА может образовываться и другими путями.
Реакция карбоксилирования ацетил-КоА является лимитирующей для всего процесса синтеза жирных кислот и может ускоряться аллостерическими положительными модуляторами: цитратом, изоцитратом и а-кетоглутаратом.
Цитоплазматический ацетил-КоА, необходимый для синтеза малонил-КоА, ведет свое происхождение от внутримитохондриального ацетил-КоА. Последний образуется в митохондриях при окислительном декарбоксилировании пирувата и ß-окислении жирных кислот. Покольку ацетил-КоА не способен проходить через митохондриальную мембрану в цитоплазму, то его переносчиком служит цитрат, который легко транспортируется через мембрану митохондрий при помощи системы переноса. Цитрат образуется путем переноса ацетил-КоА на оксалоацетат в митохондриях. После выхода в цитоплазму он вновь расщепляется АТФ-зависимым ферментом на ацетил-КоА и оксалоацетат. Отсюда становится понятной роль цитрата как положительного эффектора, стимулирующего образование малонил-КоА. Ацильные группы могут переноситься и карнитином, однако этот путь не является главным.
Далее ацетил-КоА и малонил-КоА вступают в реакцию с ацил-переносящим белком (АПБ):
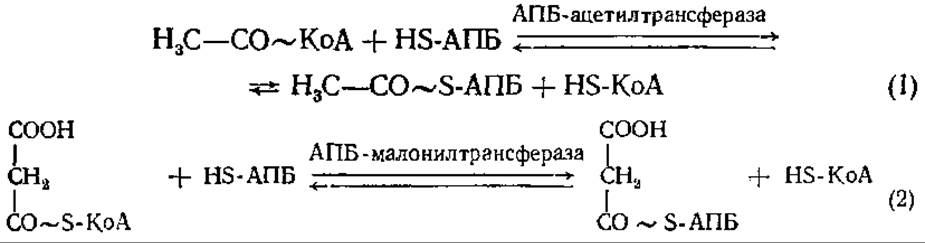
Присоединение ацильных остатков, как и в начале процесса, происходит к SH-группе 4-фосфопантетеина ацилпереносящего белка. Ацетил-S-АПБ и малонил-S-АПБ реагируют друг с другом с образованием ацетоацетил-S-АПБ, при этом из свободной СООН- группы малонил-S-АПБ (углерод отмечен звездочкой) высвобождается СO2:

Таким образом, при биосинтезе жирных кислот происходит чередование карбоксилирования и декарбоксилирования. СO2 в каждом цикле реакций внедряется в ацетил с образованием малонила и вновь выщепляется при присоединении малонила к синтезируемой цепи жирной кислоты. Смысл этой реакции заключается в том, что присоединение малонил-группировки с одновременным отщеплением СО2 идет с выделением энергии, поэтому протекает активно, не требуя дополнительных условий. Присоединение ацетил- группировок без предварительного их карбоксилирования потребовало бы потребления энергии, сопряжения с реакцией, при которой энергия выделяется. Следующие за 3-й реакцией восстановление, дегидратация и второе восстановление приводят к образованию бутирил-S-АПБ, углеродная цепочка которого на два углеродных атома длиннее исходной.

Затем реакции с 3-й по 6-ю повторяются и цепь удлиняется на два углеродных атома в каждом обороте цикла. Таким образом, для синтеза пальмитата требуется 7 циклов. Завершается синтез пальмитата отщеплением HS-АПБ от ацил-АПБ в результате гидролитического действия пальмитоил-АПБ — деацилазы. Пальмитоил-КоА ингибирует пальмитатсинтетазу, вызывая ее диссоциацию на две субъединицы, не обладающие активностью. Напротив, НАДФН активирует фермент, способствуя ассоциации субъединиц. Образование пальмитата гарантируется и тем, что деацилаза (конечный фермент процесса) способна специфически отщеплять от АПБ только пальмитат.
Ферментативные реакции синтеза пальмитиновой кислоты в известной мере представляют собой обращение процесса ее ß-окисления (реакции 4,5,6). Эти два пути метаболизма отличаются локализацией, формой переносчика ацила, а также формой, в которой добавляются или удаляются двухуглеродные единицы, специфичностью пиридиннуклеотида, стереоконфигурацией 3-гидроксиацильного промежуточного продукта, природой донора и акцептора электронов на кротонил-бутирильной стадии. Все это обеспечивает одновременное протекание в клетке противоположных процессов.
Человек и высшие животные содержат два основных источника пополнения пула жирных кислот: биосинтез пальмитиновой кислоты и поступление разнообразных жирных кислот с пищей. В печени из смеси находящихся там жирных кислот образуется тот их набор, который свойствен данному виду животного, хотя некоторое влияние на окончательный состав оказывает и диета.
Пальмитиновая и стеариновая кислоты служат предшественниками моноеновых (мононенасыщенных) жирных кислот (например, олеиновой). Введение двойной связи может происходить аэробным и анаэробным путями. У млекопитающих этот процесс осуществляется в результате переноса отщепляемого водорода на кислород с помощью ферментов десатураз (десатурация — получение ненасыщенного продукта), требующих для своего функционирования НАДН, О2 и участок микросомальной системы переноса электронов.
Особый интерес представляет вопрос о биосинтезе полиненасыщенных жирных кислот. Полиеновые кислоты, у которых двойная связь расположена между седьмым атомом углерода (отсчитывая от метила) и карбоксилом, могут образовываться из олеиновой кислоты путем чередования реакций десатурации и удлинения цепи. Животные и человек не способны к новообразованию тех полиненасыщенных жирных кислот, у которых двойные связи (или хотя бы одна из них) расположены между конечным метилом и седьмым углеродом. Эти полиненасыщенные жирные кислоты являются незаменимыми в пищевом рационе.
Разветвленные жирные кислоты синтезируются из продуктов деградации аминокислот с разветвленной цепью (валин, изолейцин и лейцин) через ацильные производные с КоА путем удлинения цепи и при участии АПБ.
Синтез триацилглицеринов. Свободные жирные кислоты не встречаются в значительных количествах в животных организмах. Они присутствуют обычно в виде сложных эфиров глицерина. Синтез триацилглицеринов происходит главным образом в печени и жировой ткани из КоА-производных жирных кислот через фосфатидную кислоту по реакции
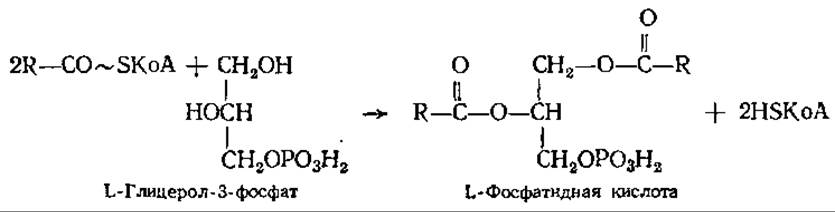
Фосфорилирование глицерина осуществляется глицеролкиназой за счет АТФ. Глицерол-3-фосфат может образовываться и при восстановлении диоксиацетонфосфата.
Гидролиз фосфатидной кислоты фосфатазой приводит к образованию 1,2-диацилглицерина, который, реагируя с другой молекулой ацил-КоА, образует нейтральный триацилглицерин.
В слизистой оболочке кишечника триацилглицерины синтезируются из свободных жирных кислот, моно- и диацилглицеринов. Эта реакция свойственна только слизистой оболочке кишечника. Перенос остатка жирной кислоты происходит через ацильное производное КоА.
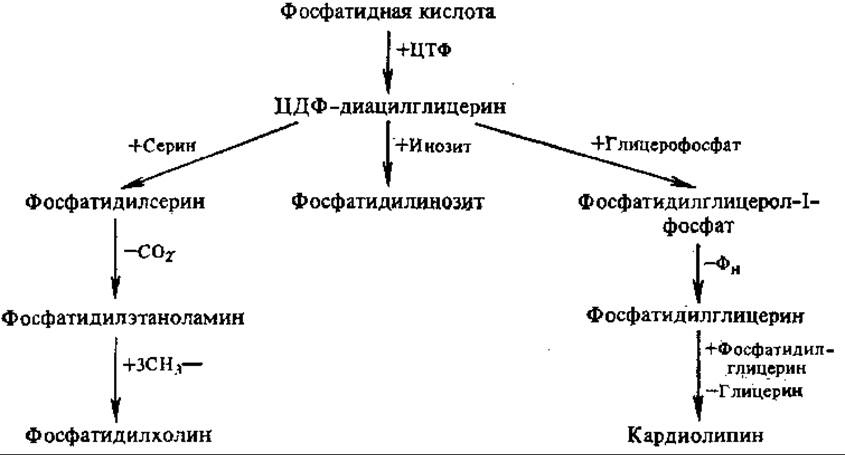
Образование глицерофосфолипидов. Предшественником для синтеза важнейших глицерофосфолипидов, как и триацилглицеринов, является фосфатидная кислота.
Биосинтез терпенов и стеринов. Хотя терпены имеют изопреновую природу, т. е. структурным звеном их молекулы является изопрен, как таковой он не участвует в биосинтезе терпенов. Исходным веществом для их синтеза служит мевалоновая кислота (3,5-дигидрокси-3-метилвалериановая):
![]()
Ацетоацетил-КоА, присоединяя еще одну молекулу ацетил-КоА и восстанавливаясь, превращается в мевалоновую кислоту. Далее в результате отщепления СО2 и активации путем образования пирофосфорного эфира при взаимодействии с АТФ мевалоновая кислота образует активные пятиуглеродные изопреноподобные звенья, конденсация которых через пирофосфорилазную реакцию приводит к наращиванию углеродной цепи, образованию различных терпенов. Конденсация шести таких пятиуглеродных звеньев дает сквален — углеводород с открытой цепью, состоящий из шести изопреновых остатков. Молекула сквалена в результате нескольких электронных сдвигов циклизуется, образуя ланостерин, а затем холестерин, который в свою очередь является предшественником многих стероидов.
