БИОХИМИЯ УЧЕБНИК ДЛЯ ВУЗОВ - Е. С. Северина - 2004
РАЗДЕЛ 8. ОБМЕН ЛИПИДОВ
V. Обмен жирных кислот и кетоновых тел
Жирные кислоты поступают с пищей или синтезируются в организме (кроме полиеновых кислот). Субстраты, необходимые для синтеза жирных кислот, образуются при катаболизме глюкозы и таким образом, часть глюкозы превращается сначала в жирные кислоты, а затем в жиры. Хотя специфический путь катаболизма жирных кислот заканчивается образованием ацетил-КоА, служащим исходным субстратом для синтеза жирных кислот, процессы синтеза и окисления жирных кислот необратимы. Они происходят в разных ком- партментах клеток (биосинтез протекает в цитозоле, а окисление — в митохондриях) и катализируются разными ферментами. Окисление жирных кислот как источников энергии увеличивается в постабсорбтивный период, при голодании и физической работе. В этих состояниях их концентрация в крови увеличивается в результате мобилизации из жировых депо, и они активно окисляются печенью, мышцами и другими тканями. При голодании часть жирных кислот в печени превращается в другие «топливные» молекулы — кетоновые тела. Они, в отличие от жирных кислот, могут использоваться нервной тканью как источник энергии. При голодании и длительной физической работе кетоновые тела служат источником энергии для мышц и некоторых других тканей.
А. β-окисление жирных кислот
β-Окисление — специфический путь катаболизма жирных кислот, при котором от карбоксильного конца жирной кислоты последовательно отделяется по 2 атома углерода в виде ацетил-КоА. Метаболический путь — β-окисление — назван так потому, что реакции окисления жирной кислоты происходят у β-углеродного атома. Реакции β-окисления и последующего окисления ацетил-КоА в ЦТК служат одним из основных источников энергии для синтеза АТФ по механизму окислительного фосфорилирования. β-Окисление жирных кислот происходит только в аэробных условиях.
Активация жирных кислот
Перед тем, как вступить в различные реакции, жирные кислоты должны быть активированы, т. е. связаны макроэргической связью с коферментом А:
RСООН + HSКоА + АТФ —> RСО ~ КоА + АМФ + РРi
Реакцию катализирует фермент ацил-КоА синтетаза. Выделившийся в ходе реакции пирофосфат гидролизуется ферментом пирофосфатазой: Н4Р2O7 + Н2O —> 2 Н3РO4.
Выделение энергии при гидролизе макроэргической связи пирофосфата смещает равновесие реакции вправо и обеспечивает полноту протекания реакции активации.
Ацил-КоА синтетазы находятся как в цитозоле, так и в матриксе митохондрий. Эти ферменты отличаются по специфичности к жирным кислотам с различной длиной углеводородной цепи. Жирные кислоты с короткой и средней длиной цепи (от 4 до 12 атомов углерода) могут проникать в матрикс митохондрий путём диффузии. Активация этих жирных кислот происходит в матриксе митохондрий. Жирные кислоты с длинной цепью, которые преобладают в организме человека (от 12 до 20 атомов углерода), активируются ацил-КоА синтетазами, расположенными на внешней мембране митохондрий.
Транспорт жирных кислот с длинной углеводородной цепью в митохондриях
β-Окисление жирных кислот происходит в матриксе митохондрий, поэтому после активации жирные кислоты должны транспортироваться внутрь митохондрий. Жирные кислоты с длинной углеводородной цепью переносятся через плотную внутреннюю мембрану митохондрий с помощью карнитина. Карнитин поступает с пищей или синтезируется из незаменимых аминокислот лизина и метионина. В реакциях синтеза карнитина участвует витамин С (аскорбиновая кислота).
В наружной мембране митохондрий находится фермент карнитинацилтрансфераза I (карнитин- пальмитоилтрансфераза I), катализирующий реакцию с образованием ацилкарнитина.
Образовавшийся ацилкарнитин проходит через межмембранное пространство к наружной стороне внутренней мембраны и транспортируется с помощью карнитинацилкарнитинтранслоказы на внутреннюю поверхность внутренней мембраны митохондрий, где фермент карнитинацилтрансфераза II катализирует перенос ацила на внутримитохондриальный КоА (рис. 8-26). Таким образом, ацил-КоА становится доступным для ферментов β-окисления. Свободный карнитин возвращается на цитозольную сторону внутренней мембраны митохондрий той же транслоказой.
Рис. 8-26. Перенос жирных кислот с длинным углеводородным радикалом через мембраны митохондрий. Фермент карнитинацилтрансфераза I — регуляторный фермент β-окисления; ингибируется малонил-КоА — промежуточным метаболитом, образующимся при биосинтезе жирных кислот. * — карнитинацилкарнитинтранслоказа.

На внутренней поверхности внутренней мембраны находится фермент карнитинацил трансфераза II, катализирующий обратный перенос ацила с карнитина на внутримитохондриальный КоА. После этого ацил-КоА включается в реакции β-окисления.
β-Окисление жирных кислот — специфический путь катаболизма жирных кислот, протекающий в матриксе митохондрий только в аэробных условиях и заканчивающийся образованием ацетил-КоА. Водород из реакций р-окисления поступает в ЦПЭ, а ацетил-КоА окисляется в цитратном цикле, также поставляющем водород для ЦПЭ. Поэтому р-окисление жирных кислот — важнейший метаболический путь, обеспечивающий синтез АТФ в дыхательной цепи.
β-Окисление начинается с дегидрирования ацил-КоА FАD-зависимой ацил-КоА дегидрогеназой с образованием двойной связи между α- и β-атомами углерода в продукте реакции — еноил-КоА. Восстановленный в этой реакции кофермент FАDН2 передаёт атомы водорода в ЦПЭ на кофермент Q. В результате синтезируются 2 молекулы АТФ (рис. 8-27). В следующей реакции р-окисления по месту двойной связи присоединяется молекула воды таким образом, что ОН-группа находится у β-углеродного атома ацила, образуя β-гидроксиацил-КоА. Затем β-гидроксиацил-КоА окисляется NAD+-зaвиcимой дегидрогеназой. Восстановленный NADH, окисляясь в ЦПЭ, обеспечивает энергией синтез 3 молекул АТФ. Образовавшийся β-кетоацил-КоА подвергается тиолитическому расщеплению ферментом тиолазой, так как по месту разрыва связи С — С через атом серы присоединяется молекула кофермента А. В результате этой последовательности из 4 реакций от ацил- КоА отделяется двухуглеродный остаток — ацетил-КоА. Жирная кислота, укороченная на 2 атома углерода, опять проходит реакции дегидрирования, гидратации, дегидрирования, отщепления ацетил-КоА. Эту последовательность реакций обычно называют «циклом β-окисления», имея в виду, что одни и те же реакции повторяются с радикалом жирной кислоты до тех пор, пока вся кислота не превратится в ацетильные остатки.
Рис. 8-27. β-Окисление жирных кислот.
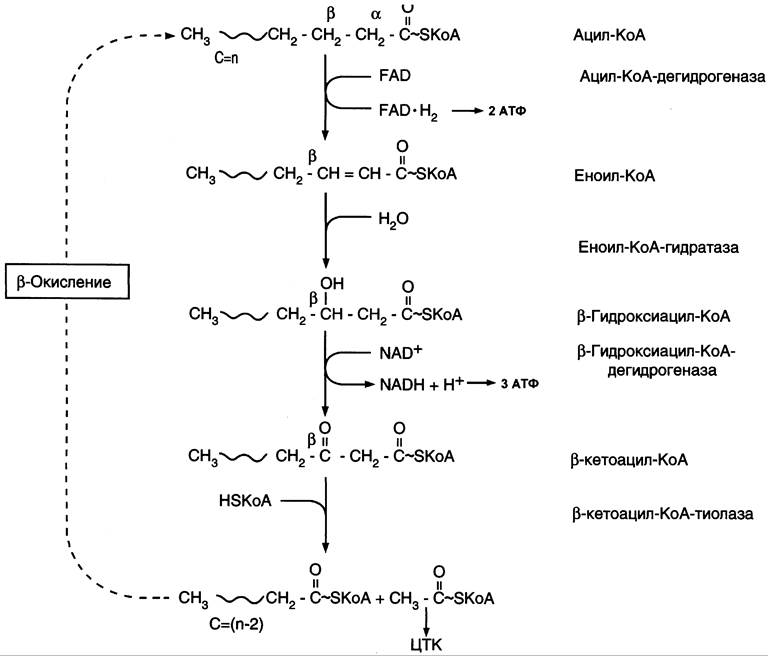
Продуктами каждого цикла β-окисления являются FADH2, NADH и ацетил-КоА. Хотя реакции в каждом «цикле» одни и те же, остаток кислоты, который входит в каждый последующий цикл, короче на 2 углеродных атома. В последнем цикле окисляется жирная кислота из 4 атомов углерода, поэтому образуются 2 молекулы ацетил-КоА, а не 1, как в предыдущих. Суммарное уравнение β-окисления, например, пальмитоил-КоА может быть представлено таким образом: С15Н31СО-КоА + 7 FAD + 7 NAD+ + 7 HSKoA —>8 СН3-СО-КoА + 7 FADH2 + 7 (NADH + H+).
Если рассчитывать выход АТФ при окислении пальмитиновой кислоты (табл. 8-7), то из общей суммы молекул АТФ необходимо вычесть 2 молекулы, так как на активацию жирной кислоты тратится энергия 2 макроэргических связей (см. реакцию активации жирной кислоты).
Таблица 8-7. Синтез АТФ при полном окислении пальмитиновой кислоты
β-Окисление |
Количество молекул АТФ |
7 NАDН (от пальмитоил-КоА до ацетил-КоА), окисление каждой молекулы кофермента в ЦПЭ обеспечивает синтез 3 молекул АТФ |
21 |
7 FАDH2, окисление каждой молекулы кофермента в ЦПЭ обеспечивает синтез 2 молекул АТФ |
14 |
Окисление каждой из 8 молекул ацетил-КоА в ЦТК обеспечивает синтез 12 молекул АТФ |
96 |
Суммарное количество молекул АТФ, синтезированных при окислении одной молекулы пальмитоил-КоА |
131 |
Во многих тканях окисление жирных кислот — важный источник энергии. Это ткани с высокой активностью ферментов ЦТК и дыхательной цепи — клетки красных скелетных мышц, сердечная мышца, почки. Эритроциты, в которых отсутствуют митохондрии, не могут окислять жирные кислоты. Жирные кислоты не служат источником энергии для мозга и других нервных тканей, так как жирные кислоты не проходят через гематоэнцефалический барьер, как и другие гидрофобные вещества. В экспериментах показано, что скорость обмена жирных кислот в нервной ткани существенно меньше, чем в других тканях.
Регуляция скорости β-окисления
β-Окисление — метаболический путь, прочно связанный с работой ЦПЭ и общего пути катаболизма. Поэтому его скорость регулируется потребностью клетки в энергии, т. е. соотношениями АТФ/АДФ и NАDН/NАD+, так же, как и скорость реакций ЦПЭ и общего пути катаболизма (см. раздел 6). Скорость β-окисления в тканях зависит от доступности субстрата, т. е. от количества жирных кислот, поступающих в митохондрии. Концентрация свободных жирных кислот в крови повышается при активации липолиза в жировой ткани при голодании под действием глюкагона и при физической работе под действием адреналина. В этих условиях жирные кислоты становятся преимущественным источником энергии для мышц и печени, так как в результате β-окисления образуются NADH и ацетил-КоА, ингибирующие пируватдегидрогеназный комплекс. Превращение пирувата, образующегося из глюкозы, в ацетил-КоА замедляется. Накапливаются промежуточные метаболиты гликолиза и, в частности, глюкозо-6-фосфат. Глюкозо-6-фосфат ингибирует гексокиназу и, следовательно, препятствует использованию глюкозы в процессе гликолиза. Таким образом, преимущественное использование жирных кислот как основного источника энергии в мышечной ткани и печени сберегает глюкозу для нервной ткани и эритроцитов.
Скорость β-окисления зависит также от активности фермента карнитинацилтрансферазы I. В печени этот фермент ингибируется малонил- КоА, веществом, образующимся при биосинтезе жирных кислот. В абсорбтивный период в печени активируется гликолиз и увеличивается образование ацетил-КоА из пирувата. Первая реакция синтеза жирных кислот — превращение ацетил-КоА в малонил-КоА. Малонил-КоА ингибирует (3-окисление жирных кислот, которые могут использоваться для синтеза жира.
Окисление ненасыщенных жирных кислот
Около половины жирных кислот в организме человека ненасыщенные. β-Окисление этих кислот идёт обычным путём до тех пор, пока двойная связь не окажется между третьим и четвёртым атомами углерода (рис. 8-28). Затем фермент еноил-КоА изомераза перемещает двойную связь из положения 3 — 4 в положение 2 — 3 и изменяет цис-конформацию двойной связи на транс-, которая требуется для β-окисления. В этом цикле β-окисления первая реакция дегидрирования не происходит, так как двойная связь в радикале жирной кислоты уже имеется. Далее циклы (3-окисления продолжаются, не отличаясь от обычного пути.
Рис. 8-28. Окисление жирных кислот с одной двойной связью.

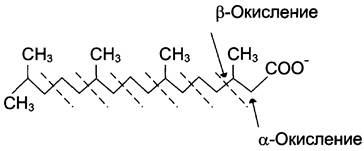
α-Окисление жирных кислот. В липидах мозга и других отделах нервной ткани преобладают жирные кислоты с очень длинной цепью — более 20 углеродных атомов. Они окисляются по типу α-окисления, при котором от жирной кислоты отщепляется по одному атому углерода, выделяющемуся в виде СO2 (рис. 8-29).
Рис. 8-29. α-Окисление жирных кислот:

Этот путь катаболизма жирных кислот не связан с синтезом АТФ. α-Окислению подвергаются также жирные кислоты с разветвлённой углеводородной цепью, например, фитановая, поступающая в организм с растительной пищей (рис. 8- 30). Фитановая кислота образуется из фитола, который входит в состав хлорофилла. В этой кислоте у каждого третьего атома углерода находится метальная группа, что делает невозможным β- окисление данной кислоты. При α-окислении фитановой кислоты вначале удаляется метальная группа, а затем происходит цикл β-окисления.
Рис. 8-30. Окисление фитановой кислоты.
Б. Нарушения окисления жирных кислот
Нарушение переноса жирных кислот в митохондрии. Скорость переноса жирных кислот внутрь митохондрий, а, следовательно, и скорость процесса β-окисления, зависит от доступности карнитина и скорости работы фермента карнитинацилтрансферазы I. β-Окисление могут нарушать следующие факторы:
✵ длительный гемодиализ, в ходе которого организм теряет карнитин;
✵ длительная ацидурия, при которой карнитин выводится как основание с органическими кислотами;
✵ лечение больных сахарным диабетом препаратами сульфонилмочевины, ингибирующими карнитинацилтрансферазу I;
✵ низкая активность ферментов, синтезирующих карнитин;
✵ наследственные дефекты карнитинацил- трансферазы I.
У людей с наследственными дефектами карнитанацилтрансферазы I или ферментов синтеза карнитина в скелетных мышцах снижается скорость поступления жирных кислот в матрикс митохондрий и, соответственно, скорость β-окисления. В этих случаях жирные кислоты с длинной цепью не используются как источники энергии. У таких людей снижена способность к физической активности; в мышечных клетках могут накапливаться жиры, образуя вакуоли.
Генетический дефект дегидрогеназы жирных кислот со средней длиной углеводородной цепи
В митохондриях имеется 3 вида ацил-КоА-дегидрогеназ, окисляющих жирные кислоты с длинной, средней или короткой цепью радикала. Жирные кислоты по мере укорочения радикала в процессе β-окисления могут последовательно окисляться этими ферментами. Генетический дефект дегидрогеназы жирных кислот со средней длиной радикала наиболее распространён по сравнению с другими наследственными заболеваниями — 1:15 000. Частота дефектного гена среди европейской популяции — 1:40. Это аутосомнорецессивное заболевание, возникающее в результате замены Т на А в 985-й позиции гена. Активность этой дегидрогеназы особенно важна для грудных детей, у которых жиры молока служат основным источником энергии, а в триацилглицеролах молока преобладают жирные кислоты со средней длиной цепи. Невозможность использовать жирные кислоты как источники энергии приводит к увеличению скорости окисления глюкозы. В результате у детей развивается гипогликемия — причина внезапной детской смертности (10% от общего числа умерших новорождённых). Если такие дети выживают, то после голодания в течение 6 — 8 ч у них развиваются гипогликемические приступы (слабость, головокружение, рвота, потеря сознания). Введение глюкозы приводит к исчезновению симптомов.
Во всех случаях, когда нарушается β-окисление, жирные кислоты накапливаются в клетках и распадаются по пути ω-окисления, которое в норме идёт с очень низкой скоростью. Окисление происходит по метальному ω-атому углерода (рис. 8-31), и в результате образуются дикарбоновые кислоты, выделяющиеся с мочой. Определение этих кислот в моче может служить диагностическим признаком нарушения β-окисления.
Рис. 8-31. ω-Окисление жирных кислот. ω-Окисление жирных кислот активируется в тех случаях, когда активность β-окисления жирных кислот снижена. 1 — адипиновая кислота; 2 — субериновая кислота.

Нарушение окисления фитановой кислоты. При редком наследственном заболевании — болезни Рефсума, развивающейся вследствие генетического дефекта одного из ферментов, участвующих в α-окислении, фитановая кислота, поступающая с пищей, не окисляется и накапливается в организме, в основном в нервной ткани. Это приводит к нарушению структуры нервной ткани и развитию многих неврологических симптомов.
В. Обмен кетоновых тел
При голодании, длительной физической работе и в случаях, когда клетки не получают достаточного количества глюкозы, жирные кислоты используются многими тканями как основной источник энергии. В отличие от других тканей мозг и другие отделы нервной ткани практически не используют жирные кислоты в качестве источника энергии. В печени часть жирных кислот превращается в кетоновые тела, которые окисляются мозгом, нервной тканью, мышцами, обеспечивая достаточное количество энергии для синтеза АТФ и уменьшая потребление глюкозы. К кетоновым телам относят β-гидроксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел.
Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления (рис. 8-32). Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил- КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.
Рис. 8-32. Активация синтеза кетоновых тел при голодании. Точечные линии — скорость метаболических путей снижена; сплошные линии — скорость метаболических путей повышена. При голодании в результате действия гпюкагона активируются липолиз в жировой ткани и β-окисление в печени. Количество оксалоацетата в митохондриях уменьшается, так как он, восстановившись до малата, выходит в цитозоль, где опять превращается в оксалоацетат и используется в глюконеогенезе. В результате скорость реакций ЦТК снижается и, соответственно, замедляется окисление ацетил-КоА. Концентрация ацетил-КоА в митохондриях увеличивается, и активируется синтез кетоновых тел. Синтез кетоновых тел увеличивается также при сахарном диабете (см. раздел 11).

Синтез кетоновых тел начинается с взаимодействия двух молекул ацетил-КоА, которые под действием фермента тиолазы образуют ацетоацетил-КоА (рис. 8-33). С ацетоацетил-КоА взаимодействует третья молекула ацетил-КоА, образуя 3-гидрокси-3-метилглутарил-КоА (ГМГ-КоА). Эту реакцию катализирует фермент ГМГ-КоА- синтаза. Далее ГМГ-КоА-лиаза катализирует расщепление ГМГ-КоА на свободный ацетоацетат и ацетил-КоА.
Рис. 8-33. Синтез кетоновых тел в митохондриях гепатоцитов. Регуляторный фермент синтеза кетоновых тел (ГМГ-КоА- синтаза) ингибируется свободным КоА. * — реакция идёт неферментативно при высокой концентрации кетоновых тел в крови.
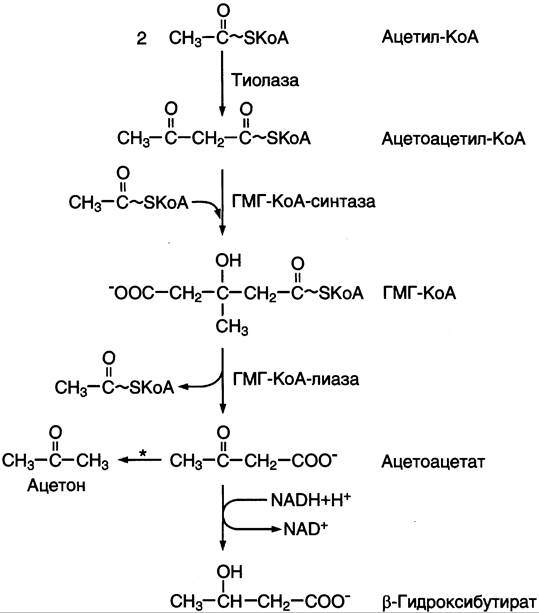
Ацетоацетат может выделяться в кровь или превращаться в печени в другое кетоновое тело — β-гидроксибутират путём восстановления.
В клетках печени при активном β-окислении создаётся высокая концентрация NADH. Это способствует превращению большей части ацетоацетата в β-гидроксибутират, поэтому основное кетоновое тело в крови — именно β-гидроксибутират. При голодании для многих тканей жирные кислоты и кетоновые тела становятся основными топливными молекулами. Глюкоза используется в первую очередь нервной тканью и эритроцитами.
При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путём организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, но, являясь водорастворимыми кислотами, вызывают ацидоз.
Регуляция синтеза кетоновых тел. Регуляторный фермент синтеза кетоновых тел — ГМГ-КоА синтаза.
✵ ГМГ-КоА-синтаза — индуцируемый фермент; его синтез увеличивается при повышении концентрации жирных кислот в крови. Концентрация жирных кислот в крови увеличивается при мобилизации жиров из жировой ткани под действием глюкагона, адреналина, т. е. при голодании или физической работе.
✵ ГМГ-КоА-синтаза ингибируется высокими концентрациями свободного кофермента А.
✵ Когда поступление жирных кислот в клетки печени увеличивается, КоА связывается с ними, концентрация свободного КоА снижается, и фермент становится активным.
✵ Если поступление жирных кислот в клетки печени уменьшается, то, соответственно, увеличивается концентрация свободного КоА, ингибирующего фермент. Следовательно, скорость синтеза кетоновых тел в печени зависит от поступления жирных кислот.
Окисление кетоновых тел в периферических тканях
При длительном голодании кетоновые тела становятся основным источником энергии для скелетных мышц, сердца и почек. Таким образом глюкоза сохраняется для окисления в мозге и эритроцитах. Уже через 2-3 дня после начала голодания концентрация кетоновых тел в крови достаточна для того, чтобы они проходили в клетки мозга и окислялись, снижая его потребности в глюкозе.
β-Гидроксибутират (рис. 8-34), попадая в клетки, дегидрируется NАБ-зависимой дегидрогеназой и превращается в ацетоацетат. Ацетоацетат активируется, взаимодействуя с сукцинил-КоА — донором КоА:
Ацетоацетат + Сукцинил-КоА —> Ацетоацетил- КоА + Сукцинат.
Рис. 8-34. Окисление кетоновых тел в тканях. * — реакция активации ацетоацетата.
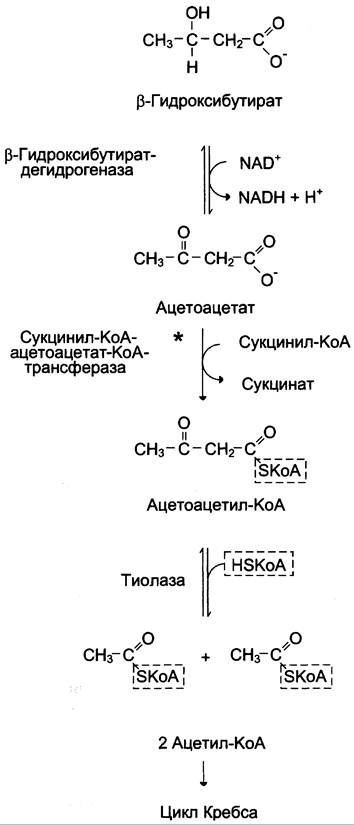
Реакцию катализирует сукцинил-КоА-ацето- ацетат-КоА-трансфераза. Этот фермент не синтезируется в печени, поэтому печень не использует кетоновые тела как источники энергии, а производит их «на экспорт». Кетоновые тела — хорошие топливные молекулы; окисление одной молекулы β-гидроксибутирата до СO2 и Н2O обеспечивает синтез 27 молекул АТФ. Эквивалент одной макроэргической связи АТФ (в молекуле сукцинил-КоА) используется на активацию ацетоацетата, поэтому суммарный выход АТФ при окислении одной молекулы β-гидроксибутирата — 26 молекул.
Кетоацидоз. В норме концентрация кетоновых тел в крови составляет 1 — 3 мг/дл (до 0,2 мМ/л), но при голодании значительно увеличивается. Увеличение концентрации кетоновых тел в крови называют кетонемией, выделение кетоновых тел с мочой — кетонурией. Накопление кетоновых тел в организме приводит к кетоацидозу: уменьшению щелочного резерва (компенсированному ацидозу), а в тяжёлых случаях — к сдвигу pH (некомпенсированному ацидозу), так как кетоновые тела (кроме ацетона) являются водорастворимыми органическими кислотами (рК~3,5), способными к диссоциации:
СН3-СО-СН2-СООН <-> СН3-СО-СН2-СОО- + H+.
Ацидоз достигает опасных величин при сахарном диабете, так как концентрация кетоновых тел при этом заболевании может доходить до 400 — 500 мг/дл. Тяжёлая форма ацидоза — одна из основных причин смерти при сахарном диабете. Накопление протонов в крови нарушает связывание кислорода гемоглобином, влияет на ионизацию функциональных групп белков, нарушая их конформацию и функцию.
Г. Биосинтез жирных кислот
С пищей в организм поступают разнообразные жирные кислоты, в том числе и незаменимые. Значительная часть заменимых жирных кислот синтезируется в печени, в меньшей степени — в жировой ткани и лактирующей молочной железе. Источником углерода для синтеза жирных кислот служит ацетил-КоА, образующийся при распаде глюкозы в абсорбтивном периоде. Таким образом, избыток углеводов, поступающих в организм, трансформируется в жирные кислоты, а затем в жиры.
1. Синтез пальмитиновой кислоты
Образование ацетил-КоА и его транспорт в цитозоль
Синтез жирных кислот происходит в абсорбтивный период. Активный гликолиз и последующее окислительное декарбоксилирование пирувата способствуют увеличению концентрации ацетил-КоА в матриксе митохондрий. Так как синтез жирных кислот происходит в цитозоле клеток, то ацетил-КоА должен быть транспортирован через внутреннюю мембрану митохондрий в цитозоль. Однако внутренняя мембрана митохондрий непроницаема для ацетил-КоА, поэтому в матриксе митохондрий ацетил-КоА конденсируется с оксалоацетатом с образованием цитрата при участии цитратсинтазы: Ацетил-КоА + Оксалоацетат —> Цитрат + НS-КоА.
Затем транслоказа переносит цитрат в цитоплазму (рис. 8-35).
Рис. 8-35. Перенос ацетильных остатков из митохондрий в цитозоль. Действующие ферменты: 1 — цитратсинтаза; 2 — транслоказа; 3 — цитратлиаза; 4 — малатдегидрогеназа; 5 — малик-фермент.
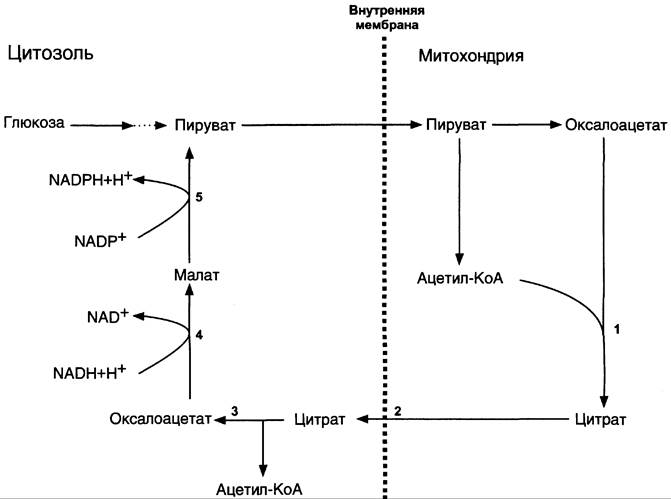
Перенос цитрата в цитоплазму происходит только при увеличении количества цитрата в митохондриях, когда изоцитратдегидрогеназа и α-кетоглутаратдегидрогеназа ингибированы высокими концентрациями NАDН и АТФ. Эта ситуация создаётся в абсорбтивном периоде, когда клетка печени получает достаточное количество источников энергии. В цитоплазме цитрат расщепляется под действием фермента цитрат- лиазы:
Цитрат + НSКоА + АТФ —> Ацетил-КоА + АДФ + Рi + Оксалоацетат.
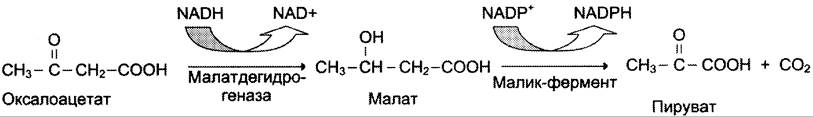
Ацетил-КоА в цитоплазме служит исходным субстратом для синтеза жирных кислот, а оксалоацетат в цитозоле подвергается следующим превращениям (см. схему ниже).
Пируват транспортируется обратно в матрикс митохондрий. Восстановленный в результате действия малик-фермента NADPH используется как донор водорода для последующих реакций синтеза жирных кислот. Другой источник NАDРН — окислительные стадии пентозофосфатного пути катаболизма глюкозы.
Образование малонил-КоА из ацетил-КоА — регуляторная реакция в биосинтезе жирных кислот.
Схема
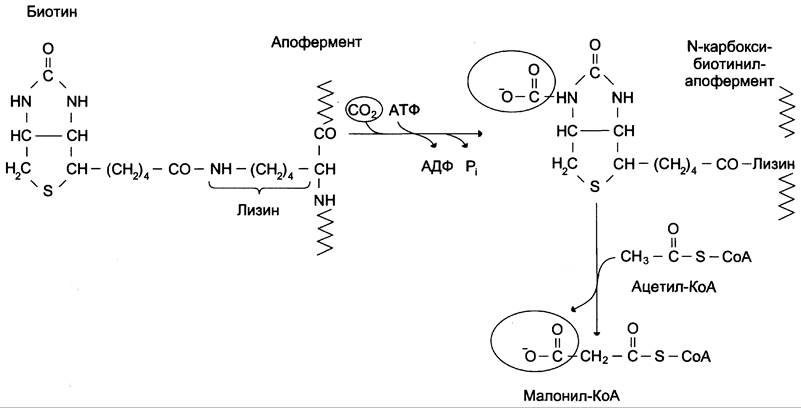
Первая реакция синтеза жирных кислот — превращение ацетил-КоА в малонил-КоА. Фермент, катализирующий эту реакцию (ацетил-КоА-карбоксилаза), относят к классу лигаз. Он содержит ковалентно связанный биотин (рис. 8-36). В первой стадии реакции СO2 ковалентно связывается с биотином за счёт энергии АТФ, во второй стадии СОО-переносится на ацетил-КоА с образованием малонил-КоА Активность фермента ацетил-КоА-карбоксилазы определяет скорость всех последующих реакций синтеза жирных кислот.
Рис. 8-36. Роль биотина в реакции карбоксилирования ацетил-КоА.
Реакции, катализируемые синтазой жирных кислот, — ферментным комплексом, катализирующим реакции синтеза пальмитиновой кислоты, описывается ниже.
После образования малонил-КоА синтез жирных кислот продолжается на мультиферментном комплексе — синтазе жирных кислот (пальмитоилсинтетазе). Этот фермент состоит из 2 идентичных протомеров, каждый из которых имеет доменное строение и, соответственно, 7 центров, обладающих разными каталитическими активностями (рис. 8-37). Этот комплекс последовательно удлиняет радикал жирной кислоты на 2 углеродных атома, донором которых служит малонил-КоА. Конечный продукт работы этого комплекса — пальмитиновая кислота, поэтому прежнее название этого фермента — пальмитоилсинтетаза.
Рис. 8-37. Строение мультиферментного комплекса — синтазы жирных кислот. Комплекс — димер из двух идентичных полипептидных цепей, каждый из которых имеет 7 активных центров и ацилпереносящий белок (АПБ). SН-группы протомеров принадлежат различным радикалам. Одна SН-группа принадлежит цистеину, другая — остатку фосфопантетеиновой кислоты. SН-группа цистеина одного мономера расположена рядом с SН-группой 4-фосфопантетеината другого протомера. Таким образом, протомеры фермента расположены «голова к хвосту». Хотя каждый мономер содержит все каталитические центры, функционально активен комплекс из 2 протомеров. Поэтому реально синтезируются одновременно 2 жирных кислоты. Для упрощения в схемах обычно изображают последовательность реакций при синтезе одной молекулы кислоты.

Первая реакция — перенос ацетильной группы ацетил-КоА на тиоловую группу цистеина ацетилтрансацилазным центром (рис. 8-38). Затем от малонил-КоА остаток малонила переносится на сульфгидрильную группу ацилпереносящего белка малонилтрансацилазным центром. После этого комплекс готов к первому циклу синтеза.
Ацетильная группа конденсируется с остатком малонила по месту отделившегося СO2. Реакция катализируется кетоацилсинтазным центром. Образовавшийся радикал ацетоацетила последовательно восстанавливается кетоацил- редуктазой, затем дегидратируется и опять восстанавливается еноилредуктазой — активными центрами комплекса. В результате первого цикла реакций образуется радикал бутирила, связанный с субъединицей синтазы жирных кислот.
Рис. 8-38. Синтез пальмитиновой кислоты. Синтаза жирных кислот: в первом протомере SН-группа принадлежит цистеину, во втором — фосфопантетеину. После окончания первого цикла радикал бутирила переносится на SН-группу первого протомера. Затем повторяется та же последовательность реакций, что и в первом цикле. Пальмитоил-Е — остаток пальмитиновой кислоты, связанный с синтазой жирных кислот. В синтезированной жирной кислоте только 2 дистальных атома углерода, обозначенные *, происходят из ацетил-КоА, остальные — из малонил-КоА.
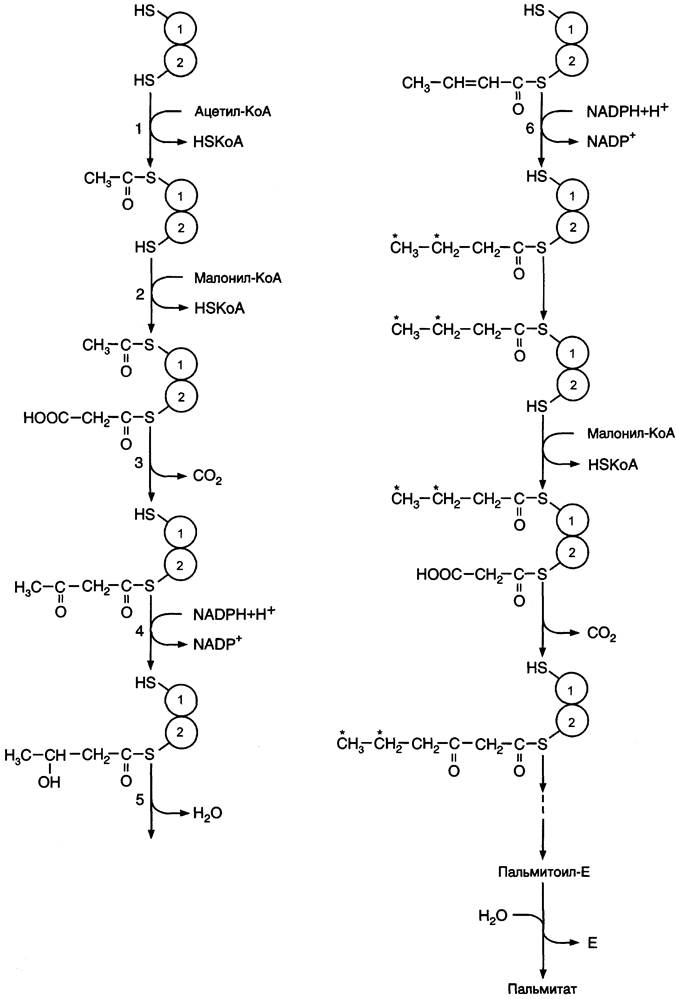
Перед вторым циклом радикал бутирила переносится из позиции 2 в позицию 1 (где находился ацетил в начале первого цикла реакций). Затем остаток бутирила подвергается тем же превращениям и удлиняется на 2 углеродных атома, происходящих из малонил-КоА.
Аналогичные циклы реакций повторяются до тех пор, пока не образуется радикал пальмитиновой кислоты, который под действием тиоэстеразного центра гидролитически отделяется от ферментного комплекса, превращаясь в свободную пальмитиновую кислоту (пальмитат, рис. 8-38, 8-39).
Рис. 8-39. Общая схема реакций синтеза пальмитиновой кислоты.
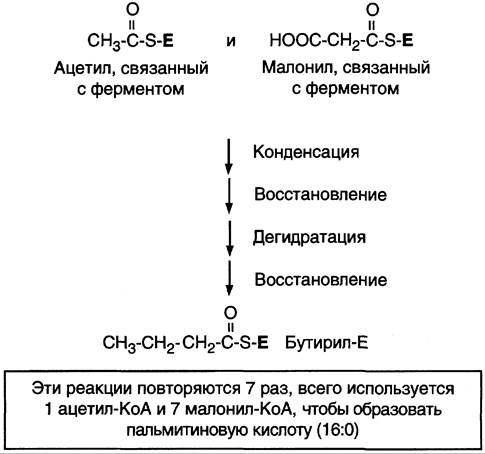
Суммарное уравнение синтеза пальмитиновой кислоты из ацетил-КоА и малонил-КоА имеет следующий вид:
СН3-СО-SКоА + 7 НООС-СН2-СО-SКоА + 14 (NАDРН + Н+) —> С15Н31СООН + 7 СO2 + 6 Н2O + 8 HSКоА + 14 NADP+.
Основные источники водорода для синтеза жирных кислот
В каждом цикле биосинтеза пальмитиновой кислоты проходят 2 реакции восстановления, донором водорода в которых служит кофермент NАDРН. Восстановление NАDР+происходит в реакциях:
✵ дегидрирования в окислительных стадиях пентозофосфатного пути катаболизма глюкозы;
✵ дегидрирования малата малик-ферментом;
✵ дегидрирования изоцитрата цитозольной NАDР-зависимой дегидрогеназой.
2. Регуляция синтеза жирных кислот
Регуляторный фермент синтеза жирных кислот — ацетил-КоА-карбоксилаза. Этот фермент регулируется несколькими способами.
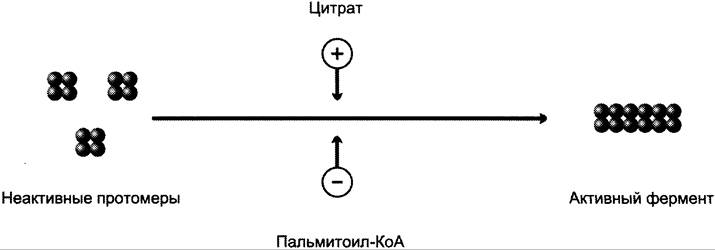
Ассоциация/диссоциация комплексов субъединиц фермента. В неактивной форме ацетил-КоА- карбоксилаза представляет собой отдельные комплексы, каждый из которых состоит из 4 субъединиц. Активатор фермента — цитрат; он стимулирует объединение комплексов, в результате чего активность фермента увеличивается. Ингибитор — пальмитоил-КоА; он вызывает диссоциацию комплекса и снижение активности фермента (рис. 8-40).
Рис. 8-40. Ассоциация/диссоциация комплексов ацетил-КоА-карбоксилазы.
Фосфорилирование/дефосфорилирование ацетил - КоА- карбоксилазы. В постабсорбтивном состоянии или при физической работе глюкагон или адреналин через аденилатциклазную систему активируют протеинкиназу А и стимулируют фосфорилирование субъединиц ацетил-КоА карбоксилазы. Фосфорилированный фермент неактивен, и синтез жирных кислот останавливается. В абсорб- тивный период инсулин активирует фосфатазу, и ацетил-КоА карбоксилаза переходит в дефосфорилированное состояние (рис. 8- 41). Затем под действием цитрата происходит полимеризация протомеров фермента, и он становится активным. Кроме активации фермента, цитрат выполняет и другую функцию в синтезе жирных кислот. В абсорбтивный период в митохондриях клеток печени накапливается цитрат, в составе которого остаток ацетила транспортируется в цитозоль.
Рис. 8-41. Регуляция ацетил-КоА-карбоксилазы.

Индукция синтеза ферментов. Длительное потребление богатой углеводами и бедной жирами пищи приводит к увеличению секреции инсулина, который стимулирует индукцию синтеза ферментов: ацетил-КоА- карбоксилазы, синтазы жирных кислот, цитратлиазы, изоцитратдегидрогеназы. Следовательно, избыточное потребление углеводов приводит к ускорению превращения продуктов катаболизма глюкозы в жиры. Голодание или богатая жирами пища приводит к снижению синтеза ферментов и, соответственно, жиров.
3. Синтез жирных кислот из пальмитиновой кислоты
Удлинение жирных кислот. В ЭР происходит удлинение пальмитиновой кислоты с участием малонил-КоА. Последовательность реакций сходна с той, что происходит при синтезе пальмитиновой кислоты, однако в данном случае жирные кислоты связаны не с синтазой жирных кислот, а с КоА. Ферменты, участвующие в элонгации, могут использовать в качестве субстратов не только пальмитиновую, но и другие жирные кислоты (рис. 8-42), поэтому в организме могут синтезироваться не только стеариновая кислота, но и жирные кислоты с большим числом атомов углерода.
Рис. 8-42. Удлинение пальмитиновой кислоты в ЭР. Радикал пальмитиновой кислоты удлиняется на 2 углеродных атома, донором которых служит малонил-КоА.

Основной продукт элонгации в печени — стеариновая кислота (С 18:0), однако в ткани мозга образуется большое количество жирных кислот с более длинной цепью — от С20до С24, которые необходимы для образования сфинголипидов и гликолипидов.
В нервной ткани происходит синтез и других жирных кислот — α-гидроксикислот. Оксидазы со смешанными функциями гидроксилируют С22 и С24 кислоты с образованием лигноцериновой и цереброновой кислот, обнаруживаемых только в липидах мозга.
Образование двойных связей в радикалах жирных кислот. Включение двойных связей в радикалы жирных кислот называется десатурацией. Основные жирные кислоты, образующиеся в организме человека в результате десатурации (рис. 8-43), — пальмитоолеиновая (С16:1∆9) и олеиновая (С18:1∆9).
Рис. 8-43. Образование ненасыщенных жирных кислот.
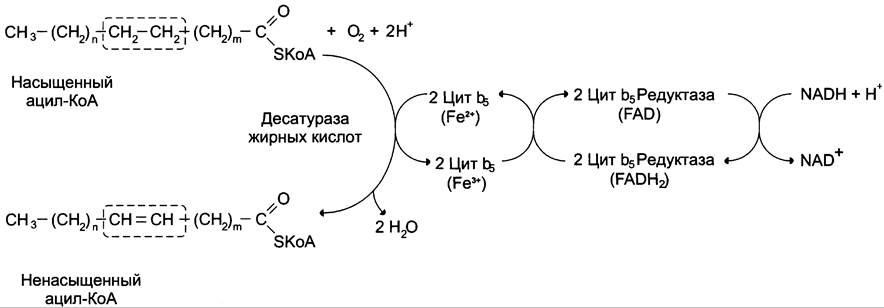
Образование двойных связей в радикалах жирных кислот происходит в ЭР в реакциях с участием молекулярного кислорода, NАDН и цитохрома b5. Ферменты десатуразы жирных кислот, имеющиеся в организме человека, не могут образовывать двойные связи в радикалах жирных кислот дистальнее девятого атома углерода, т. е. между девятым и метальным атомами углерода. Поэтому жирные кислоты семейства ω-3 и ω-6 не синтезируются в организме, являются незаменимыми и обязательно должны поступать с пищей, так как выполняют важные регуляторные функции.
Для образования двойной связи в радикале жирной кислоты требуется молекулярный кислород, NАDН, цитохром b5 и FАD-зависимая редуктаза цитохрома b5. Атомы водорода, отщепляемые от насыщенной кислоты, выделяются в виде воды. Один атом молекулярного кислорода включается в молекулу воды, а другой также восстанавливается до воды с участием электронов NАDН, которые передаются через FАDН2 и цитохром b5.