Підручник - БІОЛОГІЧНА ХІМІЯ - Губський Ю.І. - 2000
Розділ III. МЕТАБОЛІЗМ ОСНОВНИХ КЛАСІВ БІОМОЛЕКУЛ
ГЛАВА 15. МЕТАБОЛІЗМ ЛІПІДІВ. II. БІОСИНТЕЗ ТРИАЦИЛГЛІЦЕРОЛІВ І СКЛАДНИХ ЛІПІДІВ
15.4. МЕТАБОЛІЗМ СФІНГОЛІПІДІВ. СФІНГОЛІПІДОЗИ
Сфінголіпіди — складні ліпіди біологічних мембран, що побудовані на основі високомолекулярного спирту сфінгозину. Ці ліпіди — сфінгомієліни та глікосфінголіпіди в найбільшій кількості наявні в структурах центральної та периферичної нервової системи, зокрема в мієлінових оболонках нервів.
![]()
У даному розділі будуть висвітлені основні біохімічні етапи утворення сфінголіпідів та їх молекулярні порушення (ензимопатії), що мають надзвичайно важливе клінічне значення.
Біосинтез сфінголіпідів
Утворення сфінгозину
Для синтезу високомолекулярного аліфатичного аміноспирту сфінгозину використовуються вуглеводневий радикал пальмітату й залишок амінокислоти серину. Реакція каталізується ферментом, залежним від піридоксальфосфату (вітаміну В6), і потребує дії НАДФН-залежної дегідрогенази; дигідросфінгозин, що утворюється, окислюється до сфінгозину за участю ФАД-залежного флавопротеїну:
Утворення N-ацилсфінгозинів (церамідів)
Цераміди є базовою молекулярною структурою всіх сфінголіпідів. Вони утворюються шляхом N-ацилювання аміногрупи сфінгозину певною високомолекулярною жирною кислотою:
![]()
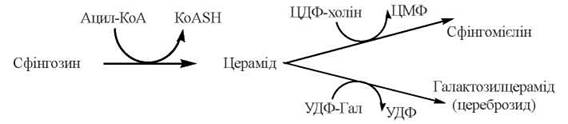
Утворення сфінгомієлінів
Сфінгомієліни — молекулярні структури, що утворюються шляхом приєднання фосфохоліну до церамідів, які містять у своєму складі залишки різних жирних кислот. Донором фосфохоліну є ЦДФ-холін:
![]()
Утворення глікосфінголіпідів
Глікосфінголіпіди — глікозильовані похідні церамідів, які, залежно від будови вуглеводної частини молекули, поділяються на: цереброзиди (моногексозиди церамідів), сульфатиди (сульфатовані похідні галактоцерамідів), глобозиди (олігогексозиди церамідів) та гангліозиди (олігосахаридні похідні церамідів, що містять у своєму складу N-ацетилнейрамінову кислоту) (глава 5). Основною біологічною функцією глікосфінголіпідів як компонентів зовнішнього шару плазматичних мембран є участь у міжклітинних взаємодіях і контактах.
Формування молекул глікосфінголіпідів здійснюється шляхом послідовного нарощування залишків моносахаридів та їх похідних на СН2ОН-групи церамідів (N-ацилсфінгозинів).
Донорами вуглеводів у цих реакціях є нуклеотидцукри; ферменти, що каталізують глікозилювання церамідів — глікозилтрансферази мембран ендоплазматичного ретикулума й апарату Гольджі. Наприклад, утворення галактозилцераміду (галактоцереброзиду) відбувається згідно з реакцією (глава 13, розділ 13.4):
![]()
Послідовність реакцій біосинтезу сфінголіпідів можна подати у вигляді такої схеми:
Катаболізм сфінголіпідів
Катаболізм сфінголіпідів здійснюється шляхом послідовного розщеплення їх молекул за участю лізосомальних гідролаз.
1. Сфінгомієліни розщеплюються до цераміду та фосфохоліну за участю сфінгомієлінази:
![]()
2. Глікосфінголіпіди. Розщеплення глікосфінголіпідів починається із поступового відщеплення моносахаридних залишків від олігосахаридного кінця молекул.
Схему гідролітичного розщеплення глікосфінголіпідів GM2 та GM1 подано на рис. 15.4. Конкретні послідовності реакцій можуть відрізнятися, залежно від особливостей будови олігосахаридної частини глікосфінголіпіду. Сфінгозин, що утворюється в результаті цих реакцій, розпадається до фосфоетаноламіну та довголанцюгового альдегіду.
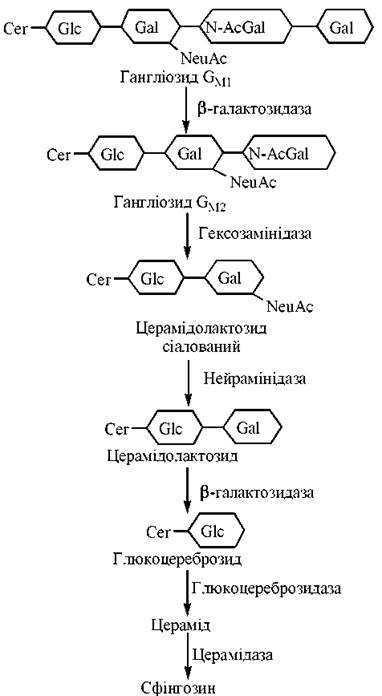
Рис. 15.4. Послідовність етапів катаболізму глікосфінголіпідів.
На схемі подані сіаловані гангліозиди (тобто такі, що зв’язані з N-ацетилнейраміновою кислотою — NeuAc).
Генетичні аномалії метаболізму сфінголіпідів
Спадкові хвороби, пов’язані з аномальним накопиченням в головному мозку та інших тканинах сфінголіпідів та продуктів їх метаболізму, структурним компонентом яких є цераміди, отримали назву сфінголіпідозів. Сфінголіпідози є ензимопатіями з класу ліпідозів, що, подібно до глікозидозів (глава 13), належать до «лізосомальних хвороб», спричинених генетичними дефектами в синтезі певних гідролітичних ферментів катаболізму складних біомолекул.
Найбільш поширеними є такі сфінголіпідози:
Хвороба Німана-Піка — сфінголіпідоз, спричинений порушенням синтезу сфінгомієлінази, що супроводжується накопиченням у головному мозку, селезінці та печінці хворих сфінгомієліну. Хвороба призводить до затримки психічного розвитку та смерті в ранньому дитячому віці.
Хвороба Тея-Сакса (гангліозидоз GM2) — генетична хвороба, спричинена дефектом у синтезі гексозамінідази, що відщеплює термінальний N-ацетилгалактозамін від гангліозиду GM2, який в аномальних кількостях накопичується в головному мозку. Хвороба проявляється затримкою розумового розвитку, сліпотою, неврологічними розладами, макроцефалією; смерть хворих дітей звичайно настає у віці 3-4 років. Цей сфінголіпідоз найбільш поширений серед етнічних євреїв — вихідців із Центральної та Східної Європи, де частота хвороби, зокрема в популяції єврейського населення США, досягає 1 випадку на 3600 новонароджених.
Гангліозидоз GM1 — сфінголіпідоз, що спричиняється порушенням синтезу лізосомальної β-галактозидази, яка відщеплює термінальний залишок галактози в гангліозиді GM1 і кератансульфатах (таблиця 13.3) із накопиченням у нервовій системі відповідних сполук. За клінічною картиною гангліозидоз — хвороба, близька до хвороби Тея-Сакса.
Хвороба Гоше (глюкоцереброзидний ліпідоз) — сфінголіпідоз, генетичний дефект при якому полягає в недостатньому синтезі глюкоцереброзидази — ферменту, що відщеплює залишок глюкози від молекул глюкоцереброзидів, які накопичуються в ретикулоендотеліальній системі. Патологія має поліморфну клінічну картину і проявляється спленомегалією, збільшенням печінки, ураженням кісткової тканини, нейропатіями.