Основы биохимической инженерии Часть 1 - Бейли Дж., Оллис Д. 1989
Кинетика катализируемых ферментами реакций
Другие факторы, влияющие на ферментативную активность
Скорость ферментативных реакций и температура
При изучении любых проблем химической кинетики одним из основных теоретических положений является уравнение Аррениуса, выражающее зависимость константы скорости реакция от температуры:
![]()
где Еа — энергия активации реакции; R — газовая постоянная; А — предэкспоненциальный множитель; 7 — абсолютная температура.
При графическом изображении зависимость Ink от 1/7 выражается прямой с тангенсом угла наклона — Ea/R [конечно, если соблюдается условие уравнения (3.66)]. Аррениусовской зависимости константы скорости реакции от температуры подчиняются и многие реакции, катализируемые ферментами; примером могут служить экспериментальные данные, представленные на рис. 3.24.
Следует подчеркнуть, однако, что температурный диапазон, в котором были получены приведенные на рис. 3.24 данные, очень узок. В этом эксперименте не рассматривались температуры, значительно превышающие обычные в биологических условиях пределы. Что же произойдет, если мы попытаемся заставить фермент катализировать процесс еще быстрее, подняв температуру еще выше? Как показано на рис. 3.25, в большинстве случаев результат окажется очень печальным.

РИС. 3.24. Аррениусовская зависимость скорости ферментативной реакции (катализируемый миозином гидролиз АТР) от температуры. [Воспроизведено из работы: Laidler К. J., The Chemical Kinetics of Enzyme Action, p. 197, The Clarendon Press, Oxford, 1958; данные из статьи Quellet L., Laidler K. Morales M. F., Molecular Kinetics of Muscle Adenosine Triphosphate, Arch. Biochem. Biophys., 39, 37 (1952).]
Денатурация большинства белков начинается в диапазоне температур от 45 до 50 °С и завершается очень быстро при 55°С. Один из механизмов термической денатурации белков очевиден: по мере повышения температуры атомы в молекуле белка приобретают все более высокую энергию, в том числе кинетическую, и в конце концов становится возможным разрушение слабых связей, стабилизирующих глобулярную структуру белка, что и приводит к его инактивации.
Термическая инактивация ферментов может быть обратимой, необратимой или смешанной. Зависимость скорости реакции ферментативного катализа от температуры в достаточно широком диапазоне температур может быть описана с помощью простой модели обратимой термической инактивации. Согласно этой модели, мы принимаем, что неактивная (і) и активная (а) формы фермента находятся в равновесии:
![]()
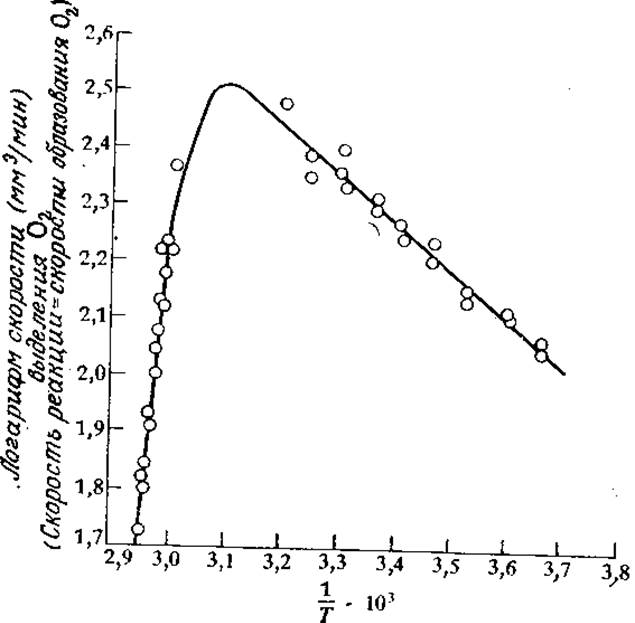
РИС. 3.25. При высоких температурах, когда начинает доминировать процесс термической инактивации фермента, аррениусовская зависимость скорости реакции от температуры нарушается (приведен пример разложения Н2О каталазой). Сплошная кривая вычислена по уравнению (3.73) при следующих значениях параметров: E = 3,5 ккал/моль, ∆Hd = 55,5 ккал/моль, ∆Sd = 168 ккал/(моль∙К), ß = 258 мм3/мин. [Воспроизведено из статьи: Sizer I.W., Temperature Activation and Inactivation of the Crystalline Catalase-Hydrogen Peroxide System, J. Biol. Chem., 154, 461 (1944).]
Константу равновесия этой реакции можно выразить следующим уравнением:
![]()
В этом уравнении символами ∆Gd, ∆Нd и ∆Sdобозначены свободная энергия, энтальпия и энтропия инактивации соответственно.
Хотя изолированные водородные связи сравнительно слабы (их энергия обычно изменяется в пределах от 3 до 7 ккал/ /моль), энтальпия инактивации ферментов ∆Нd достаточно высока, составляя, например, для трипсина и лизоцима белка куриных яиц соответственно 68 и 73,5 ккал/моль. Инактивация этих ферментов сопровождается изменением энтропии, равным + 213 кал/(моль∙К). Благодаря высокой энтальпии денатурации уже небольшие изменения температуры существенно изменяют относительное количество активной формы фермента. При таких высоких значениях ∆Нd фермент инактивируется практически полностью в диапазоне тридцати градусов.
Поскольку весь фермент существует либо в активной, либо в неактивной форме, т. е.
eа + ei = e0 (3.69)
из уравнения (3.68) следует, что
![]()
Согласно теории переходного состояния, скорость реакций (3.4) при высоких концентрациях субстрата может быть выражена следующим образом:
vmax = ea∙k(T) (3.71)
где
![]()
Здесь kB и h — константы Больцмана и Планка соответственно, а а — коэффициент. Из уравнений (3.68), (3.70) — (3.72) следует, что
![]()
В уравнении (3.73) коэффициент ß включает a, kB, h, е0 и ехр(∆S*/R).
Это уравнение и представляет собой математическое выражение, описывающее схематично изображенное на рис. 3.25 поведение систем фермент — субстрат. Показанная на этом рисунке сплошная линия, проходящая через экспериментально найденные точки, фактически была вычислена по уравнению (3.73) после определения параметров ß, Е, ∆Sd и ∆Нd на основе тех же экспериментальных данных. При больших значениях 1/Т тангенс угла наклона кривой приближенно равен —Е/R (погрешность равна Т (К) и обычно не вносит большого вклада). Тангенс угла наклона другого прямого участка кривой при более высоких температурах равен приблизительно (∆На—Е)/ R. Величину ∆Sd можно определить, используя тот факт, что при температуре Тmах, когда lnvmax принимает наивысшее значение,
![]()
[Это выражение легко получить, приняв d(lnvmax)/dT равным нулю.] Поскольку Тmax известна из измерений, а величины Е и ∆Нd мы уже определили, то не представляет труда вычислить правую часть уравнения (3.74). Теперь, зная величину Kd(Tmах), возвратимся к уравнению (3.68) и найдем ASd. Наконец, подберем такое значение коэффициента ß, чтобы vmах [Tmах в уравнении (3.73)] стало равным измеренной величине. При необходимости более точные значения параметров можно получить методом последовательного приближения. В частности, очень большое влияние на результаты может оказывать значение Тmax. Конечно, от температуры зависят и другие параметры уравнений скорости реакции, например константа Михаэлиса или ингибиторная константа. Если эти параметры считать константами равновесия, как это часто бывает в действительности, то мы должны прийти к температурной зависимости типа той, которая выражается уравнением (3.68). Тогда график зависимости lnК (или рК) от 1/T будет представлять собой прямую линию, по наклону которой можно определить стандартную свободную энергию.
В некоторых случаях графическая зависимость в этих координатах изображается не прямой линией, иными словами, наклон линии меняется при изменении диапазона температур. Причиной подобных осложнений могут быть неоправданные упрощения в интерпретации Кm или Kiили даже в последовательности катализируемых ферментом реакций. Напомним, например, что для случая простейшей последовательности реакций (3.4) Кm представляет собой не что иное, как отношение констант скоростей элементарных стадий реакции, выражаемое уравнением (3.5). Если каждую из этих констант элементарных стадий в свою очередь выразить в форме уравнения Аррениуса [уравнение (3.66)] или в форме уравнения теории переходного состояния [уравнение (3.72)], то получится весьма сложная форма зависимости от температуры. Другим возможным источником затруднений в определении зависимости некоторых параметров скорости реакции от температуры может служить существование нескольких промежуточных фермент-субстратных комплексов (или комплексов фермента с продуктом реакции).
Подобные соображения были положены в основу полезного и довольно широко применяемого способа проверки корректности кинетических моделей и соответствующих гипотетических последовательностей реакций. Если зависимость кинетических параметров в аррениусовских координатах выражается не прямой, то исходная модель или чрезмерно упрощена, или вообще неверна.