ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 2. БИОЭНЕРГЕТИКА И МЕТАБОЛИЗМ - 2014
ЧАСТЬ II. БИОЭНЕРГЕТИКА И МЕТАБОЛИЗМ
23. ГОРМОНАЛЬНАЯ РЕГУЛЯЦИЯ И ИНТЕГРАЦИЯ МЕТАБОЛИЗМА МЛЕКОПИТАЮЩИ
23.4. Ожирение и регуляция массы тела
В США 30% взрослого населения страдает ожирением, а еще у 35% избыточный вес. (Для определения ожирения рассчитывают специальный показатель — индекс массы тела (ИМТ или англ. BMI)
ИМТ = масса тела (кг) / (рост человека)2 (м)2
ИМТ |
|
В норме |
<25 |
Избыточный вес |
25-30 |
Ожирение |
>30 |
Ожирение опасно для жизни; оно значительно увеличивает вероятность развития диабета II типа, инфаркта миокарда, инсульта, а также злокачественных опухолей толстой кишки, молочной железы, простаты и эндометрия. Поэтому очень важно понимать, каким образом осуществляется регуляция массы тела и происходит накопление жиров в жировой ткани. ■
Упрощенно можно сказать, что ожирение — это результат поступления с пищей большего количества калорий, чем это было необходимо для организма. Организм может использовать лишнюю энергию тремя способами: 1) запасать в виде жиров и хранить в жировой ткани, 2) расходовать избыток энергии при экстремальных нагрузках, и 3) превратить энергию в тепло (термогенез), разобщая дыхательную цепь митохондрий. У млекопитающих целый набор сигналов от эндокринной и нервной систем контролирует баланс поступления энергии и затраты организма таким образом, что количество жировой ткани поддерживается на нужном уровне. Чтобы эффективно бороться с ожирением необходимо понимать, как это равновесие регулируется в норме и как эти механизмы гомеостаза могут нарушаться.
Жировая ткань выполняет важную эндокринную функцию
Поддержание постоянной массы тела удобно объяснять на модели механизма обратной связи в адипоцитах. Согласно этой модели, в организме действует механизм ингибирования пищевого поведения и увеличения потребления энергии, когда масса тела превышает определенную величину. При снижении масса тела до определенной величины ингибирование ослабляется (рис. 23-33). Сигнал обратной связи приходит из жировой ткани в мозговые центры, контролирующие пищевое поведение и метаболическую и двигательную активность организма. Из факторов патогенеза первым был открыт лептин в 1994 г., а последующие исследования показали, что жировая ткань служит важным эндокринным органом, где синтезируются пептидные гормоны, названные адипокинами. Адипокины могут вызывать локальный эффект (аутокринное и паракринное действие) или системный эффект (эндокринное действие), передавая информацию о состоянии энергетических резервов (триацилглицеринов) в жировой ткани другим тканям и в головной мозг.
Рис. 23-33. Модель механизма поддержания постоянной массы. При увеличении массы жировой ткани высвобождается лептин; он снижает аппетит и ингибирует синтез жиров, а также стимулирует окисление жирных кислот. При уменьшении массы жировой ткани образование лептина снижается; это приводит к тому, что пищи поглощается больше, а скорость окисления жирных кислот уменьшается.
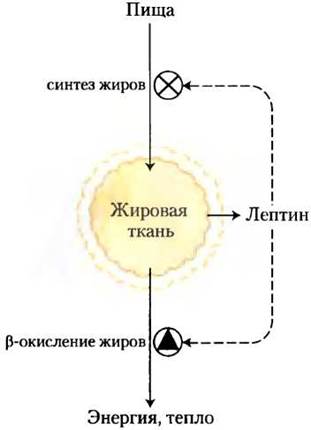
В норме адипокины регулируют изменения метаболизма топливных молекул и потребность организма в питании, благодаря чему восстанавливаются необходимые запасы энергетических веществ и поддерживается нормальная масса тела. При избытке или недостатке адипо- кинов регуляция нарушается, что может стать смертельно опасным.
Адипокин лептин (от греч. leptos — тонкий) (167 аминокислотных остатков) поступает в головной мозг, действует на рецепторы в гипоталамусе, что вызывает снижение аппетита. Лептин впервые был идентифицирован у лабораторных мышей как продукт гена ОВ (от англ. obese — тучный). У мышей с генотипом ob/ob, т. е. с двумя дефектными копиями этого гена (строчными буквами обозначают мутантную форму гена), проявляются поведенческие и физиологические реакции, характерные для животных, находящихся в состоянии постоянного голодания. У них повышен уровень кортизола в плазме крови. Они не способны сохранять тепло, у них наблюдаются отклонения в размерах, они не размножаются и обладают неограниченным аппетитом. Из-за этого у таких мышей быстро развивается сильное ожирение, масса их тела в 3 раза превышает норму (рис. 23-34). У них нарушен метаболизм; как и больные диабетом, эти мыши не чувствительны к инсулину. Если мышам с генотипом оb/оb инъецировать лептин, они теряют в весе, у них увеличивается локомоторная активность и образование тепла.
Рис. 23-34. Ожирение вызывается нарушениями в выработке лептина. Обе эти мыши одного возраста и обе дефектны по гену ОВ. Справа — мышь, ежедневно получающая инъекции очищенного лептина; масса этой мыши 35 г. Слева — мышь, не получающая лептин, поэтому она ела больше пищи, была менее активна; масса этой мыши 67 г.

Обнаружен и второй мышиный ген DВ (диабетический), который играет роль в регуляции аппетита. Мыши с двумя дефектными копиями (db/db) страдают ожирением и больны диабетом. Ген DВ кодирует рецептор лептина, а если рецептор лептина дефектен, сигнальная функция лептина теряется.
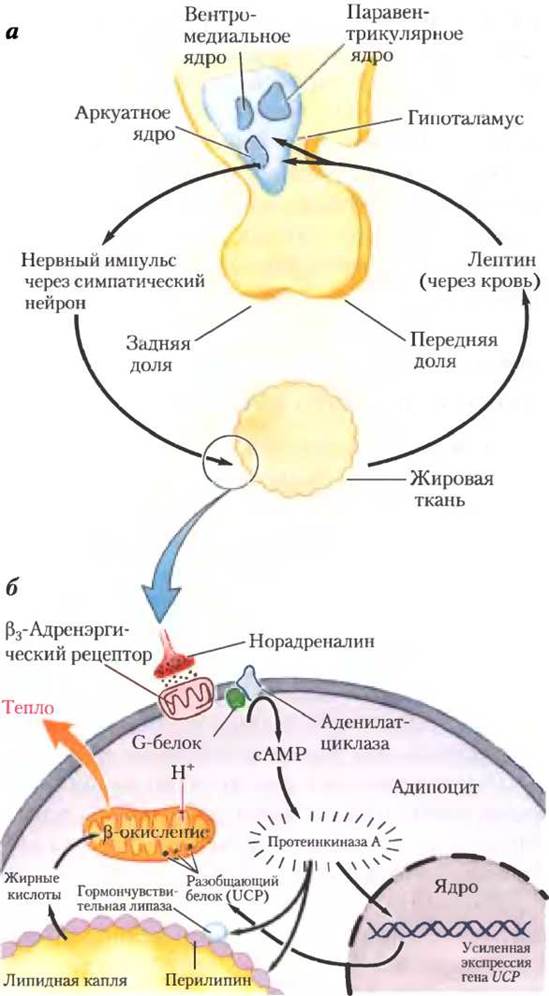
Рецептор лептина преимущественно экспрессируется в нейронах аркуатных ядер гипоталамуса — регуляторов пищевого поведения (рис. 23-35, а). Лептин передает информацию о больших запасах жира. Принятый тканями сигнал приводит к снижению потребления жира и увеличению расходования энергии. Взаимодействие лептина с рецептором в гипоталамусе изменяет нервные сигналы в зоны мозга, регулирующие аппетит. Кроме того, лептин стимулирует симпатическую нервную систему, вызывая повышение кровяного давления и частоты сердечных сокращений, а также усиливает образование тепла, разобщая в митохондриях адипоцитов процесс переноса электронов и синтез ATP (рис. 23-35, б). Вспомним, что термогенин (разобщающий белок UCP) образует канал во внутренней мембране митохондрий, что позволяет протонам вновь проникать в митохондриальный матрикс без прохождения через АТР- синтазный комплекс. Это дает возможность непрерывно продолжать окисление энергоемких молекул (жирных кислот в адипоцитах) без синтеза АТР, переводя всю энергию в тепло, и использовать энергию пищи или запасенного жира в очень больших количествах.
Рис. 23-35. Гипоталамическая регуляция потребления пищи и энергии, а — анатомия гипоталамуса. б — взаимосвязи между гипоталамусом и жировыми клетками, описано ниже в тексте.
Лептин стимулирует образование пептидных гормонов, снижающих аппетит
В аркуатном ядре гипоталамуса нейроны двух типов контролируют поглощение источников энергии и метаболизм (рис. 23-36). Орексигенные (стимулирующие аппетит) нейроны стимулируют прием пищи, вырабатывая и высвобождая нейропептид Y (NРY), в результате чего следующий в цепи нейрон посылает сигнал в мозг: «Есть!». Во время голодания уровень NPY в крови увеличивается; он повышен и у мышей с генотипом ob/оb и db/db. У мышей, у которых нет чувства насыщения, главная причина ожирения тоже связана с высоким уровнем NPY.
Рис. 23-36. Гормоны, контролирующие пищевое поведение. В аркуатном ядре гипоталамуса есть два набора нейросекреторных клеток, которые воспринимают гормональный сигнал и передают нервные импульсы клеткам мышц, жировой ткани и печени. Лептин и инсулин высвобождаются из жировой ткани и поджелудочной железы (соответственно) пропорционально массе жира в теле. Эти два гормона действуют на анорексигенные нейросекреторные клетки (красный), инициируя выделение α-МСГ; таким образом формируются сигналы «есть меньше и метаболизировать больше энергетических запасов». Лептин и инсулин действуют и на орексигенные нейросекреторные клетки (зеленый цвет), ингибируя высвобождение NPY и уменьшая сигнал «есть» тканям. Как описано в тексте, желудочный гормон грелин стимулирует аппетит, активируя экспрессирующие NPY клетки, а PYY3-36, синтезируемый в прямой кишке, ингибирует эти нейроны, и таким образом снижает аппетит. Нейросекреторные клетки любого из этих двух типов ингибируют выработку гормонов друг у друга, некоторые стимулы, активирующие орексигенные клетки, инактивируют анорексигенные клетки, и наоборот. Это усиливает эффект стимулирующих сигналов.
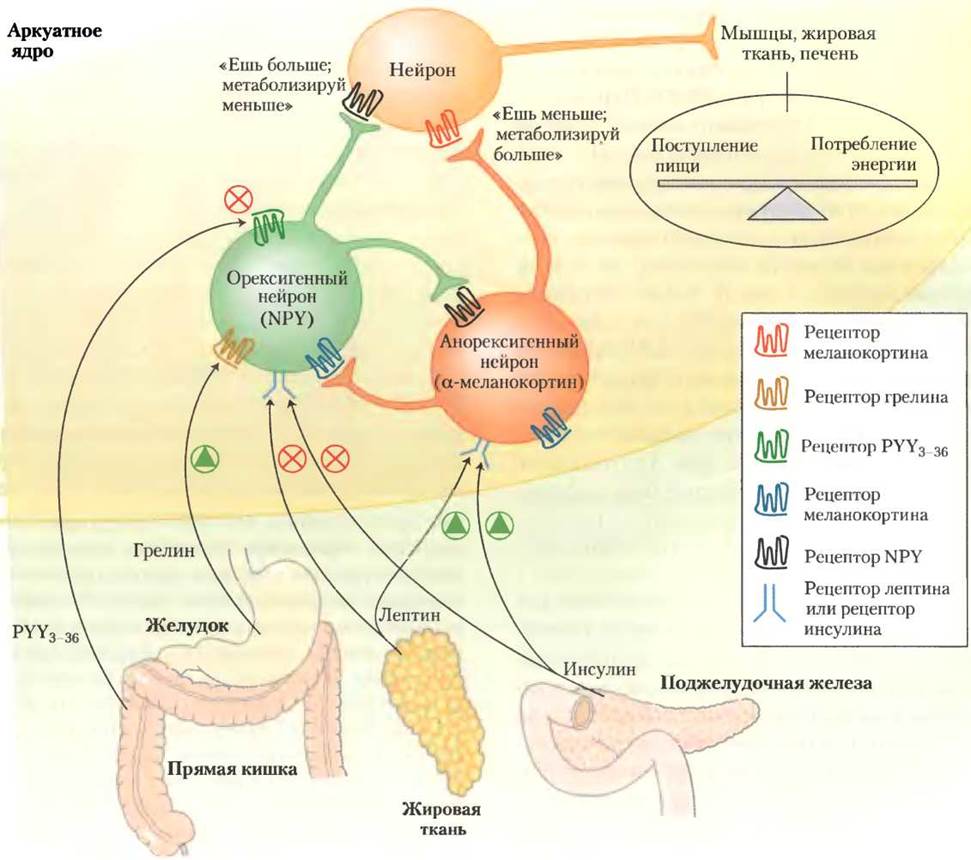
Анароксигенные (подавляющие аппетит) нейроны в аркуатном ядре гипоталамуса вырабатывают α-меланоцитстимулирующий гормон (α-МСГ, или меланокортин), образующийся из полипептидного предшественника проопиомеланокортина (ПОМК; рис. 23-6). При высвобождении α-МСГ следующий нейрон в цикле посылает в мозг сигнал: «Перестать есть!».
От количества и размеров адипоцитов зависит количество лептина, выделяемого жировой тканью. Когда в результате потери веса снижается масса липидной ткани, то и концентрация лептина в крови падает, образование NPY угнетается — процесс, показанный на рис. 23-35, обращается. В ответ на уменьшение интенсивности сАМР-зависимой сигнализации разобщение угнетается, замедляя повышение температуры; при этом жиры запасаются, а мобилизация жиров уменьшается. Употребление большего количества пищи при более эффективном усвоении жиров приводит к пополнению запасов жира, и система опять возвращается в состояние равновесия.
Кроме того, лептин, вероятно, необходим для нормального развития нервных связей в гипоталамусе. У мышей рост нервной ткани аркуатного ядра на раннем этапе формирования мозга замедляется в отсутствие лпетина; это влияет и на орексигенные, и (в меньшей степени) на анорексигенные нейроны гипоталамуса. Возможно, уровень лептина во время формирования этих связей определяет детали строения самой регуляторной системы.
Лептин включает сигнальный каскад, регулирующий экспрессию генов
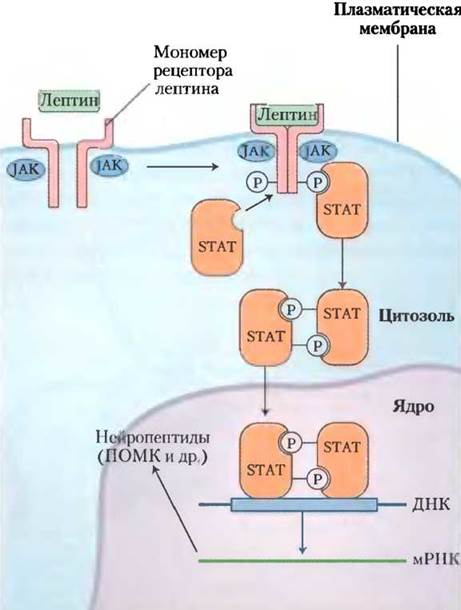
Лептиновый сигнал преобразуется с помощью JAК-SТАТ-механизма, который также используется рецептором интерферона и рецептором фактора роста (рис. 23-37; см. также рис. 12-18 вт. 1). Рецептор лептина обладает единственным трансмембранным участком. При связывании лептина с внеклеточным доменом двух мономеров рецептора происходит их димеризация. Оба мономера фосфорилируются с помощью янус-киназы (JAK) по тирозиновому остатку внутриклеточного домена. Остатки (P)-Туr становятся местами стыковки для трех белков-активаторов транскрипции (STAT 3, 5 и 6). Прикрепленные белки STAT затем фосфорилируются по остаткам Туr с помощью той же самой JAK. После фосфорилирования белки STAT димеризуются, затем входят в ядро, где связываются со специфическими последовательностями ДНК и стимулируют экспрессию генов-мишеней, в том числе и ген ПОМК, из которого образуется α-МСГ.
Рис. 23-37. Механизм JAK-STAT трансдукции сигнала лептина в гипоталамус. Связывание лептина индуцирует димеризацию рецептора лептина, после которой происходит фосфорилирование остатков Туr-рецептора, катализируемое янус-киназой (JAK). Белки STAT через свои SН2-домены связываются с фосфорилированным лептиновым рецептором, после чего фосфорилируются по остаткам Туr благодаря специфической активности JAK. Затем STAT димеризуются, связываясь друг с другом по фосфорилированным тирозиновым остаткам ((P)-Туr), и проникают в ядро. Здесь они взаимодействуют со специфическими регуляторными участками ДНК и меняют экспрессию определенных генов. В итоге продукты этих генов влияют на пищевое поведение и потребление организмом энергии.
Одна из причин усиления катаболизма и повышения тепловыделения, запускаемых лептином, — увеличение синтеза в адипоцитах белка термогенина, разобщителя дыхательной цепи митохондрий (продуцируется геном UCP1). Лептин стимулирует синтез термогенина, изменяя синаптическую передачу от нейронов в аркуатном ядре в жировую и другие ткани. В этих тканях лептин вызывает увеличение выделения норадреналина, который, взаимодействуя с β3- адренорецепторами, стимулирует транскрипцию гена UCP1. В результате происходящего разобщения электрон-транспортной цепи с окислительным фосфорилированием расходуются жиры и выделяется тепло (рис. 23-35).
Может ли ожирение быть связано с неэффективным образованием лептина? Можно ли в таком случае вылечить его инъекциями лептина? Но на самом деле концентрация лептина в крови у животных (и людей) с ожирением выше, чем у особей с нормальной массой тела (за исключением, конечно, животных с оb/оb-генотипом, которые не могут вырабатывать лептин). У ожиревших особей в системе ответа на лептин должны быть дефектными некоторые последовательные стадии, и повышение уровня лептина, по-видимому, является результатом безуспешной попытки организма вернуть чувствительность к лептину. В очень редких случаях у людей с чрезвычайно сильным ожирением дефектен ген лептина (ОВ). При этом инъекции лептина должны приводить к значительной потере веса. Но у подавляющего большинства страдающих ожирением ген ОВ не поврежден. В клинической практике инъекции лептина не оказывают на людей эффекта снижения веса, наблюдаемого у ожиревших мышей с генотипом ob/ob. Очевидно, у людей в большинстве случаев ожирения наряду с лептином существенную роль играют какие-то другие факторы.
Лептиновая система участвует в регуляции ответа на истощение
Вероятно, лептин возник в процессе эволюции для адаптации активности и метаболизма животных в периоды голода, а не как средство, ограничивающее увеличение массы тела. Снижение уровня лептина при недостаточном питании приводит к обращению процесса термогенеза (рис. 23-35) и позволяет сохранить энергетические запасы организма. Лептин (действующий в гипоталамусе) также вызывает снижение образования тиреоидных гормонов (ослабляя основной метаболизм), снижение уровня синтеза половых гормонов (предотвращая репродукцию), а также стимулирует выработку глюкокортикоидов (мобилизуя энергетические ресурсы организма). Таким образом, опосредованный лептином ответ, приводящий к снижению расхода энергии и стимуляции использования эндогенных источников энергии, позволяет организму пережить затяжные периоды голодания. В печени и мышцах лептин стимулирует АМР-активируемую протеинкиназу (АМРК), что приводит к ингибированию синтеза жирных кислот и активации окисления жирных кислот и в итоге способствует протеканию процессов получения энергии.
Инсулин действует в аркуатном ядре, регулируя питание и запасание энергии
Секреция инсулина регулирует как объем запасов жира (тучность), так текущее энергетическое состояние организма (уровень глюкозы крови). Инсулин действует на инсулиновые рецепторы в гипоталамусе, снижая аппетит (рис. 23-36). Рецепторы инсулина в орексигенных нейронах в аркуатном ядре ингибируют высвобождение NPY, а рецепторы инсулина в анорексигенных нейронах стимулируют образование α-МСГ, снижая тем самым поглощение топливных молекул и влияя на метаболизм жирных кислот и углеводов в печени и в мышцах. Адипонектин увеличивает выработку тепла. С помощью механизмов, обсуждаемых в разд. 23.3, инсулин также сигнализирует мышцам, печени и жировой ткани о необходимости увеличения интенсивности превращения глюкозы в ацетил-СоА, тем самым обеспечивая исходный материал для синтеза жиров.
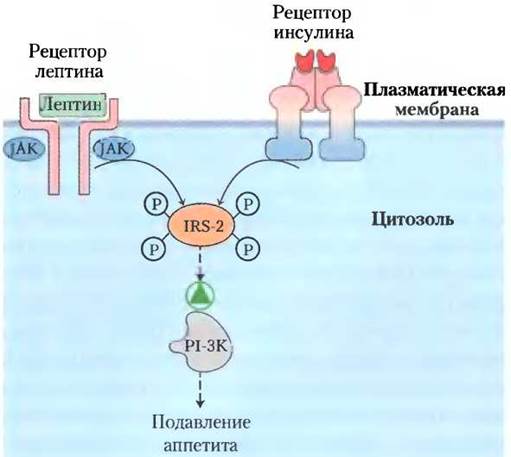
Лептин увеличивает чувствительность клеток печени и мышц к инсулину. Возможно, этот эффект обусловлен взаимодействием между протеин-тирозинкиназами, активируемыми лептином, и киназами, активируемыми инсулином (рис. 23-38); общие вторичные мессенджеры в двух сигнальных путях позволяют лептину инициировать некоторые события, которые запускаются также инсулином через комплекс инсулин-рецептор-субстрат-2 (IRS-2) и фосфоинозитид-3-киназу (РI-3К) (см. рис. 12-16 в т. 1).
Рис. 23-38. Вероятный механизм взаимодействия между рецепторами инсулина и лептина. Рецептор инсулина обладает собственной тирозинкиназной активностью (см. рис. 12-15 в т. 1), а лептиновый рецептор при связывании своего лиганда фосфорилируется растворимой формой тирозинкиназы (JAK). Одно из возможных объяснений наблюдаемого взаимодействия между лептином и инсулином состоит в том, что они оба могут фосфорилировать один и тот же субстрат; здесь это комплекс инсулин-рецептор-субстрат-2 (IRS-2). В фосфорилированном виде IRS-2 активирует PI-3K, участвующую в последовательности событий, включающей подавление аппетита. IRS-2 суммирует импульсы от обоих рецепторов.
Адипонектин увеличивает чувствительность к инсулину, действуя через АМРК
Адипонектин — пептидный гормон (224 аминокислотных остатка), синтезируемый почти исключительно в жировой ткани; этот адипокин повышает чувствительность других органов к действию инсулина, предотвращает развитие атеросклероза и ингибирует воспалительный ответ (адгезию моноцитов, трансформацию макрофа
гов, а также пролиферацию и миграцию гладкомышечных клеток сосудов). Адипонектин увеличивает поглощение миоцитами жирных кислот из крови, а также увеличивает скорость (3-окисления жирных кислот в мышцах. Он также блокирует синтез жирных кислот и глюконеогенез в гепатоцитах и стимулирует поглощение глюкозы и ее катаболизм в мышцах и печени.
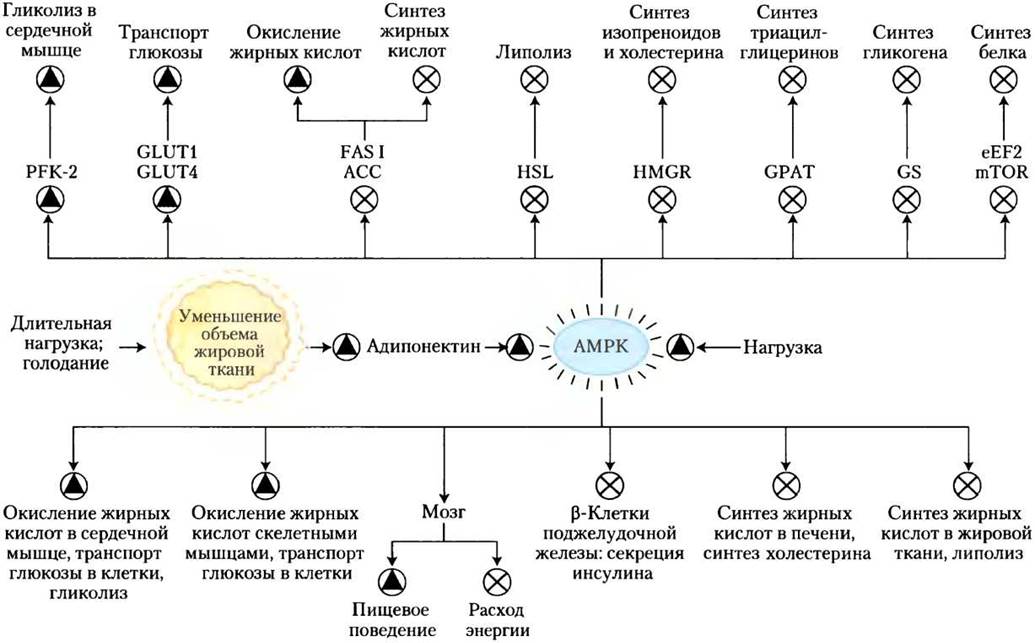
Это действие адипонектина осуществляется не непосредственно, и отдельные элементы этой системы еще предстоит изучить, однако известно, что многие из процессов происходят при участии АМР-активируемой протеинкиназы (АМРК). Вспомните (см. рис. 15-6), что АМРК активируется под действием факторов, сигнализирующих о необходимости изменить метаболизм от биосинтеза в сторону образования энергии (рис. 23-39). Активированная АМРК фосфорилирует ключевые в метаболизме липидов и углеводов белки, вызывая системные эффекты в метаболизме организма (рис. 23-40). Рецепторы адипонектина также присутствуют в головном мозге; гормон активирует АМРК в гипоталамусе, стимулируя аппетит и снижая расход энергии.
Рис. 23-39. Роль АМР-активируемой протеинкиназы (АМРК) в регуляции метаболизма АТР. Образующийся в реакциях синтеза ADP превращается в АМР под действием аденилаткиназы. АМР активирует АМРК, которая регулирует процессы анаболизма и катаболизма путем фосфорилирования ключевых ферментов (см. рис. 23-40).
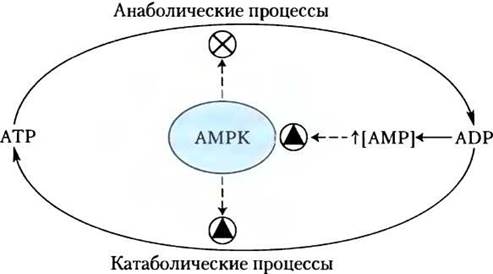
Рис. 23-40. Образование адипонектина и его действие через АМРК. При длительном голодании запасы триглицеринов в жировой ткани сокращаются, что стимулирует синтез адипонектина и его высвобождение из адипоцитов. Адипонектин плазмы крови воздействует на специфические рецепторы в плазматической мембране самых разных клеток и органов, ингибируя процессы, протекающие с большими затратами энергии, и стимулируя процессы, приводящие к накоплению энергии. В головном мозге адипонектин действует через свои рецепторы, стимулируя организм к приему пищи, ингибируя энергозатратные виды физической активности, а также ингибируя термогенез в бурой жировой ткани. Этот гормон осуществляет свое действие путем активации АМРК, которая регулирует (через фосфорилирование) специфические ферменты в ключевых метаболических процессах (см. рис. 15-6). Принятые сокращения: PFK-2 — фосфофруктокиназа-2; GLUT1 и GLUT4 — переносчики глюкозы; FASI — синтаза жирных кислот типа I; АСС — ацетил-СоА-карбоксилаза; HSL — гормончувствительная липаза; HMG-CoA-редуктаза — 3-гидрокси-3-метилглутарил- СоА-редуктаза; GPAT — глицерофосфатацилтрансфераза; GS — гликогенсинтетаза; eEF2 — эукариотический фактор элонгации 2 (необходим для синтеза белка, см. гл. 27); mTOR — мишень рапамицина у млекопитающих (протеинкиназа, регулирующая синтез белка в зависимости от доступности питательных веществ). Лекарственные препараты из группы тиазолидиндионов активируют фактор транскрипции PPARy (см. рис. 23-41 и 23-42), который включает синтез адипонектина, опосредованно активируя АМРК. Физическая нагрузка (результат — превращение АТР в ADP и АМР) также стимулирует АМРК.
Один из регулируемых АМРК ферментов в печени и белой жировой ткани — ацетил-СоА- карбоксилаза, образующая малонил-СоА, первый промежуточный продукт в биосинтезе жирных кислот. Малонил-СоА — это мощный ингибитор фермента карнитинацилтрансферазы I, начинающей процесс β-окисления, осуществляя транспорт жирных кислот в митохондрии (рис. 17-6).
Путем фосфорилирования и инактивации ацетил-СоА-карбоксилазы АМРК ингибирует синтез жирных кислот, так как ослабляет (с помощью малонил-СоА) ингибирование β-окисления (см. рис. 17-12). Синтез холестерина также ингибируется при действии АМРК, которая фосфорилирует и инактивирует НМG- СоА-редуктазу — один из ферментов реакций биосинтеза холестерина (см. рис. 21-34). Кроме того, АМРК ингибирует синтазу жирных кислот и ацилтрансферазу, эффективно блокируя синтез триглицеринов.
Мыши с дефектным геном адипонектина менее чувствительны к инсулину, чем носители гена дикого типа, и они плохо переносят повышение уровня глюкозы. Эти нарушения метаболизма имеют симптомы, очень похожи на диабет II типа (когда нарушается чувствительность к инсулину — инсулиннезависимый диабет, ИНЗД). При этом происходит также снижение уровня глюкозы крови, но только медленнее. Действительно, у людей с ожирением или у больных диабетом II типа уровень глюкозы крови ниже, чем у пациентов контрольной группы без ожирения. Более того, препараты группы тиазолидиндионов розглитазон (Avandia) и пиоглитазон (Actos), используемые при лечении диабета II типа (с. 470), повышают экспрессию мРНК адипонектина в жировой ткани и увеличивают уровни адипонектина у экспериментальных животных. Эти лекарства активируют также АМРК (рис. 23-40). (В 2007 г. была пересмотрена безопасность препарата Авандия в связи с повышенным риском инфаркта миокарда.) Вполне возможно, что адипонектин опосредует связи ожирения с диабетом II типа; ожирение — один из главных факторов риска развития диабета. ■
Экспрессия генов, играющих главную роль в поддержании массы тела, регулируется рационом питания
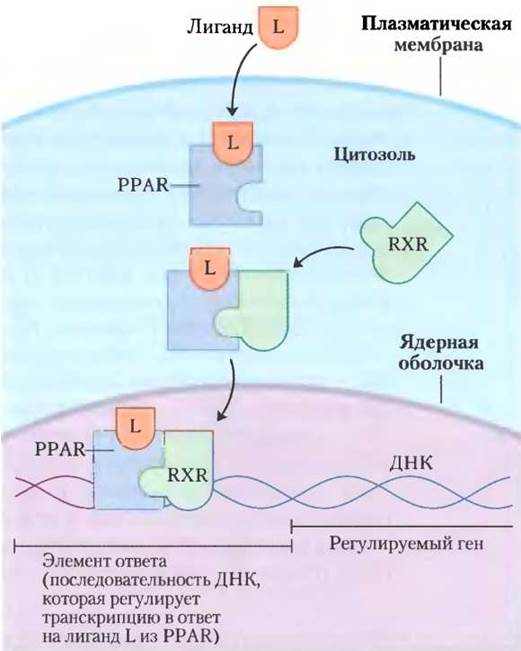
На изменения содержания липидов в диете реагирует семейство белков лиганд-активируемые факторы транскрипции, которые представлены пероксисомнымирецепторами, активируемыми пролифераторами (англ. peroxisome proliferator- activated receptors, PPARs). Они влияют на экспрессию генов, участвующих в метаболизме жиров и углеводов. Сначала была установлена роль этих факторов транскрипции при осуществлении синтезов в пероксисомах, соответственно этому они и были названы. Их обычными лигандами являются жирные кислоты или их производные, но они способны также связывать и синтетические агонисты. Эти факторы транскрипции можно активировать в лабораторных условиях с помощью генетических манипуляций. PPARα, PPARδ и PPARy входят в суперсемейство ядерных рецепторов. Они действуют в ядре, формируя гетеродимеры с другим ядерным рецептором RXR (рецептор ретиноидов X), связанным с регуляторными областями ДНК, которые расположены вблизи контролируемых ими генов и изменяют скорость транскрипции этих генов (рис. 23-41).
Рис. 23-41. Механизм действия РРАR. При связывании своих лигандов РРАR образуют гетеродимеры с ядерным рецептором RXR. Димер связывает специфические участки ДНК — элементы ответа, стимулируя транскрипцию генов в этих участках.
Рецепторы РРАRy экспрессируются в основном в печени и жировой ткани и участвуют в регуляции генов, определяющих дифференцировку фибробластов в адипоциты, и генов, кодирующих белки, необходимые для синтеза и хранения липидов в адипоцитах (рис. 23-42). РРАRy активируются препаратами группы тиазолидиндионов, которые используются для лечения диабета II типа.
Рис. 23-42. Метаболическая функция изоформ PРАR. Три изоформы РРАR регулируют гомеостаз жиров и глюкозы путем координированного влияния на экспрессию генов в печени, мышцах и жировой ткани. Формы РРАRα и РРАRδ (и близкородственная им форма РРАRβ регулируют усвоение жиров; РРАRy регулирует запасание жиров и чувствительность различных тканей к инсулину.
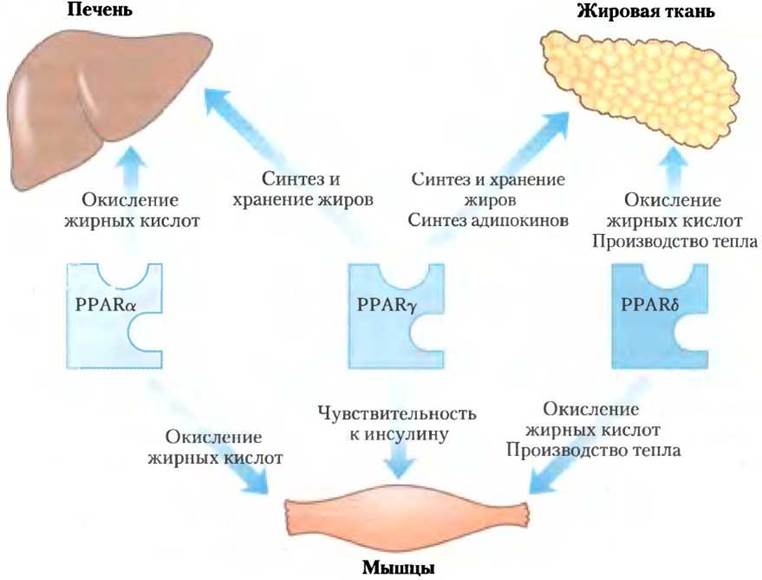
Рецепторы РРАRα экспрессируются в печени, почках, сердце, скелетных мышцах и бурой жировой ткани. К лигандам, активирующим этот фактор транскрипции, относятся эйкозаноиды, свободные жирные кислоты, а также лекарственные препараты из группы фибратов, такие как фенофибрат (Трайкор) и ципрфибрат (Модалим), которые применяются при лечении ИБС с целью повышения уровня ЛПВП и снижения уровня триглицеринов крови. В гепатоцитах РРАRα включает гены, необходимые для усвоения питательных веществ и для β-окисления жирных кислот и образования кетоновых тел во время голодания.
Рецепторы РРАRδ (и очень близкая к нему изоформа РРАRβ) — ключевые регуляторы окисления липидов, изменяющие чувствительность тканей к количеству липидов в пище. Они действуют в печени и мышцах, стимулируя транскрипцию по крайней мере девяти генов, кодирующих ферменты β-окисления и высвобождения энергии посредством разобщения дыхательной цепи митохондрий. Если нормальных мышей перекормить большим количеством жиров, они набирают много бурого и белого жира, а накопление жиров в печени у них снижается. Но когда эти же эксперименты с перееданием проводят на мышах с постоянно активным вследствие генетических изменений РРАRδ, такого накопления жиров не происходит. У мышей с дефектным лептиновым рецептором (db/db) активированный PPARδ предотвращает развитие ожирения, которое происходит, если PPARδ не активирован (рис. 23-34). Стимулируя распад жирных кислот в разобщенных дыхательных цепях митохондрий, PPARδ вызывает расщепление жиров, потерю веса и выделение тепла. С учетом вышесказанного можно сделать вывод, что выделение тепла в равной степени важно и для поддержания температуры тела, и для предупреждения ожирения. Несомненно, что при лечении ожирения рецепторы PPARδ вполне подходят как мишень для лекарственных средств.
Влияние грелина и PYY3-36 на кратковременное пищевое поведение
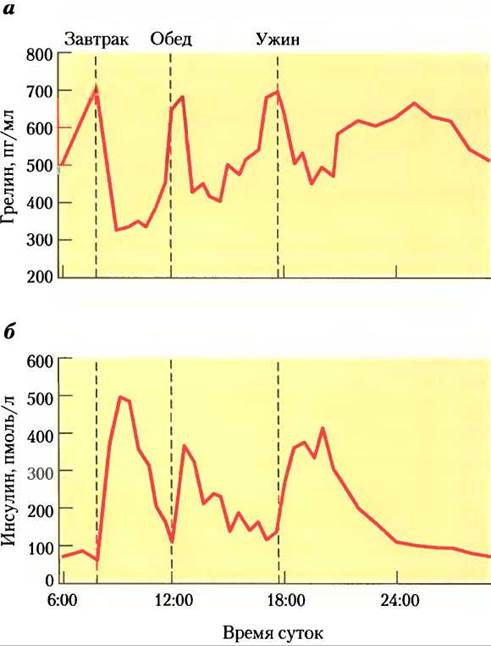
Грелин — это пептидный гормон (28 аминокислот), вырабатываемый клетками слизистой оболочки желудка. Первоначально он был обнаружен как стимулятор высвобождения гормона роста (ghre, праиндоевропейский корень ’grow’); впоследствии было показано, что он заметно повышает аппетит и работает на более коротких промежутках времени, чем лептин и инсулин (между приемами пищи). Грелиновые рецепторы расположены в гипофизе (вероятно, они опосредуют высвобождение гормона роста) и в гипоталамусе (контроль аппетита), а также в сердечной мышце и в жировой ткани. Между приемами пищи концентрация грелина в крови значительно варьирует, достигая максимума перед приемом пищи и резко падая сразу после приема пищи (рис. 23-43). Инъекции грелина немедленно вызывают у человека чувство сильного голода. При синдроме Прадера-Вилли уровень грелина крови исключительно высокий, у больных неконтролируемый аппетит, что приводит к чрезвычайному ожирению и часто является причиной преждевременной смерти до 30 лет.
Рис. 23-43. Изменения в уровнях грелина и инсулина связаны с временем приема пищи, а — концентрация грелина в плазме резко увеличивается непосредственно перед привычным временем приема пищи (завтрак 7:00, обед 12:00, ужин 17:30) и резко падает сразу после приема пищи; при этом кажется, что чувство голода нарастает. б — концентрация инсулина увеличивается сразу после каждого приема пищи, это происходит в ответ на увеличение содержания глюкозы в крови.
РYY3-36 — это пептидный гормон (34 аминокислоты), секретируемый эндокринными клетками слизистой тонкого кишечника и прямой кишки в ответ на прохождение пищи из желудка. Уровень РУУ3-36 в крови увеличивается после приема пищи и остается высоким в течение нескольких часов. С кровотоком РУУ3-36 достигает аркуатного ядра, где воздействует на орексигенные нейроны, ингибируя высвобождение NPY и притупляя чувство голода (рис. 23-36). Люди, которым инъецировали РУУ3-36, ощущают голод очень слабо, они едят меньше нормы в течение примерно 12 ч после инъекции.
Итак, нейроэндокринная система контролирует потребление пищи и метаболизм, преимущественно обеспечивая защиту от истощения и сверхпродуктивного накопления жира (экстремальное ожирение). Характерные трудности, которые испытывают многие люди, старающиеся снизить вес, свидетельствуют о высокой эффективности этих видов контроля.
Краткое содержание раздела 23.4 Ожирение и регуляия массы тела
■ Число людей, страдающих ожирением, непрерывно увеличивается, и это общая проблема развитых стран. Ожирение заметно снижает качество жизни и сопряжено с другими опасными заболеваниями.
■ Жировая ткань вырабатывает лептин — гормон, регулирующий пищевое поведение и расходование энергии так, чтобы поддерживать адекватные запасы жира. Образование лептина и его высвобождение увеличиваются прямо пропорционально количеству и размерам адипоцитов (жировых клеток).
■ Лептин действует на рецепторы в аркуатном ядре гипоталамуса, вызывая высвобождение анорексигенных пептидов, в том числе α-МСГ, который действует в мозге, подавляя аппетит. Лептин также стимулирует действие симпатической нервной системы на адипоциты, что приводит к разобщению окислительного фосфорилирования в митохондриях и последующему выделению тепла.
■ Механизм передачи сигнала лептина связан с фосфорилированием системы JAK-STAT. При фосфорилировании с помощью JAK белки STAT могут связываться с регуляторными участками ядерной ДНК и изменять экспрессию генов ферментов, которые устанавливают уровень метаболической активности и пищевое поведение. Инсулин действует на рецепторы в аркуатном ядре с таким же результатом, как и лептин.
■ Гормон адипонектин стимулирует поглощение жирных кислот и их окисление, а также ингибирование синтеза жирных кислот. Это осуществляется с помощью АМРК, которая активируется также при низкой концентрации АМР и при физических нагрузках.
■ Гормон грелин вырабатывается в желудке, а действует на орексигенные нейроны в аркуатном ядре гипоталамуса, формируя чувство голода перед приемом пищи. Пептидный гормон кишечника PYY3-36 действует там же, но он подавляет чувство голода после приема пищи.