Биохимия - Химические реакции в живой клетке Том 2 - Д. Мецлер 1980
Коферменты - особые природные специализированные реагенты
Тиаминдифосфат
Ферментативные реакции с участием тиамина
Все известные реакции, зависимые от тиаминдифосфата, могут быть составлены из пяти полуреакций (а—е на рис. 8-3), каждая из которых является а-расщеплением, приводящим к образованию связанного с тиамином «активного альдегида» (центральная часть рис. 8-3), который идентичен соединению, изображенному в левой части уравнения (8-14). Декарбоксилирование а-кетокислоты до альдегида представлено стадией б с последующей реакцией в направлении, обратном стадии а. Наиболее изученным ферментом, катализирующим реакцию этого типа, является пируватдекарбоксилаза дрожжей. Это нуждающийся в Mg2+ димерный фермент с мол. весом ~200 000 [28]. Тиаминдифосфат и Mg2+ отщепляются от фермента при pH 8, однако активный фермент можно реконструировать при pH ниже 7.
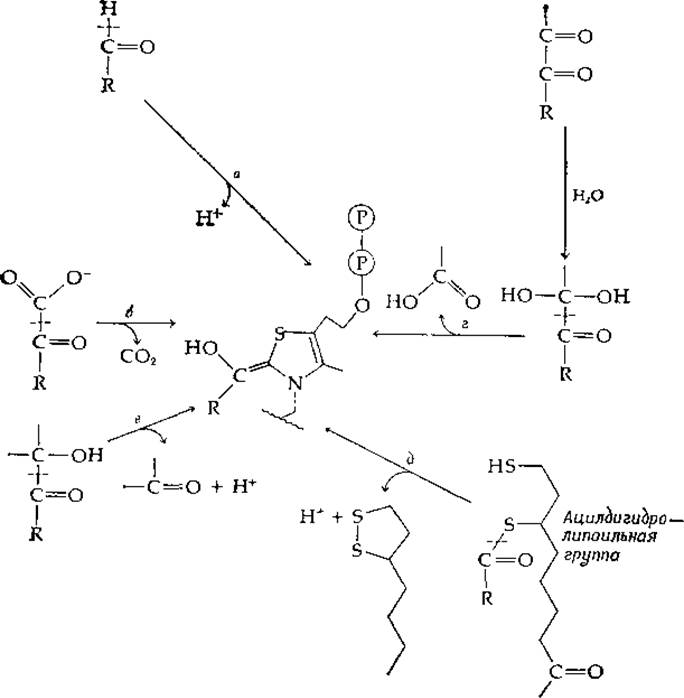
РИС 8-3. Полуреакции, составляющие тиаминзависимые реакции а-расщепления и а-конденсации (реакция типа 7 в табл. 9-1).
Образование а-кетолов из а-кетокислот также начинается со стадии б, за которой следует конденсация с другим карбонильным соединением, осуществляемая обращением стадии в. Хорошо известным примером такой реакции служит синтез а-ацетолактата

в котором сочетаются декарбоксилирование пирувата и конденсация образующегося активного ацетальдегида с другой молекулой пирувата. Эта реакция катализируется ацетолактатсинтетазой, которую иногда именуют «карболиазой». Ацетолактат служит предшественником валина и лейцина (рис. 14-10). Сходная кетольная конденсация необходима и в биосинтезе изолейцина (рис. 14-10). Ацетолактат является ß-кетокислотой и легко декарбоксилируется в ацетоин; эта реакция имеет важное значение при некоторых видах бактериального брожения [уравнение (9-28)]. Кетольная конденсация двух молекул глиоксилата с декарбоксилированием катализируется глиоксилат-карболигазой (гл. 9, разд. В, 2).
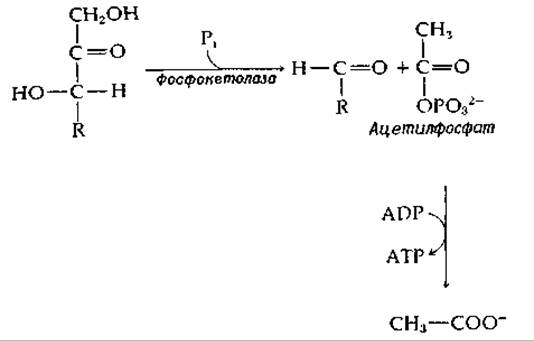
РИС. 8-4. Расщепление а-кетола до альдегида и карбоновой кислоты; последовательность реакций, сопряженная с фосфорилированием ADP (реакция типа S7A).
Другой путь ферментативного синтеза кетолов состоит в расщеплении альдегида (стадия а) с последующей конденсацией со вторым альдегидом (стадия в в обратном направлении). а-Дикетоны могут расщепляться (стадия г) на карбоновую кислоту и активный альдегид, который далее подвергается превращению в реакции, соответствующей стадии а или стадии в в обратном направлении. Эти и другие сочетания стадий часто наблюдаются как побочные реакции при действии таких ферментов, как пируватдекарбоксилаза. Родственной реакцией, зависимой от тиамина, является реакция между пируватом и ацетил-СоА, ведущая к образованию а-дикетона, диацетила СН3СОСОСН3 [29]. Эту реакцию можно рассматривать как замещение СоА-аниона из ацетил-СоА в результате атаки связанным с тиамином активным альдегидом, образовавшимся из пирувата (обращение стадии г на рис. 8-3 с высвобождением СоА).
Стадия д на рис. 8-3 представляет собой реакцию расщепления производного ацилдигидролипоата. Реакция обычно протекает в обратном направлении и является частью процесса окислительного декарбоксилирования а-кетокислоты, который начинается стадией б (см. разд. К).
Важное значение имеет реакция расщепления а-кетолов, в которой используется стадия в (рис. 8-3) с последующим обращением этой же стадии, но с другим акцептором альдегида. Эту реакцию катализирует транскетолаза [уравнение (9-15)] — фермент, необходимый в пентозо-фосфатных путях метаболизма и в фотосинтезе. Родственная реакция (рис. 8-4), которая имеет более сложный механизм, катализируется ферментом фосфокетолазой; эта реакция играет важную роль в энергетическом метаболизме некоторых бактерий. Продуктом реакции, катализируемой фосфокетолазой, является ацетилфосфат, расщепление которого может быть сопряжено с синтезом АТР (рис. 8-4).
Мы полагаем, что требуемое а-расщепление приводит к образованию тиаминсодержащего соединения (верхнее соединение на приводимой ниже схеме):
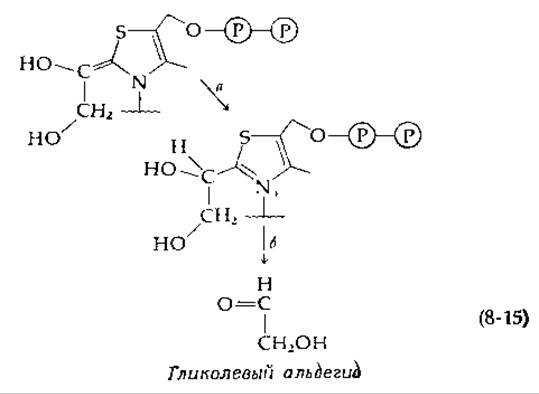
Протонирование этого промежуточного соединения (стадия а) могло бы сопровождаться распадом до альдегида гликолевой кислоты (стадия б). Обычно полагают, что синтез гликолевого альдегида может протекать в хлоропластах именно этим путем как побочная реакция, катализируемая транскетолазой (гл. 13, разд. Д, 9, б). Однако фосфокетолаза катализирует реакцию, которая в сущности является внутримолекулярной оксидоредукцией. Альдегидная группа альдегидного фрагмента гликолевой кислоты окисляется в карбоновую кислоту, в то время как —СН2ОН-группа восстанавливается до —СН3. Один из возможных механизмов включает образование 2-ацетилтиаминдифосфата в качестве промежуточного соединения, тогда как в основе другого варианта лежит присоединение Рі к двойной связи.
Дополнение 8-Г
Тиамин (витамин B1)
История открытия и выделения тиамина описана в дополнении 8-А. В 1937 г. К. Ломан и П. Шустер выделили чистую «кокарбоксилазу» — диализируемый кофермент, необходимый для декарбоксилирования пирувата при помощи фермента из дрожжей. Было показано, что им является тиаминдифосфат (рис. 8-2). Моно-, три- и тетрафосфаты также встречаются в природе, но в меньших количествах.
Физические свойства. Тиамин и его коферментные формы — белые твердые вещества, легко растворимые в воде. Тиамин обычно поступает в продажу в форме дихлорида (мол. вес 337,3), а тиаминдифосфат — в форме монохлорида (мол. вес 460,8).
Кислотно-основные реакции. В щелочном растворе тиамин претерпевает двухстадийную реакцию раскрытия тиазольного кольца с образованием аниона тиольной формы, которую можно закристаллизовать в виде натриевой соли:
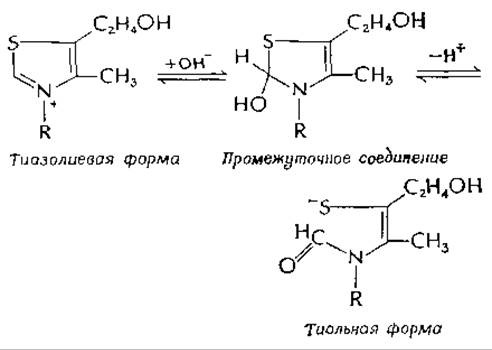
Эта реакция, аналогично параллельно протекающей реакции, описанной в гл. 4 [уравнение (4-31)], которая приводит к образованию желтой нестабильной формы тиамин-аниона, является примером практически полностью кооперативного отщепления двух протонов, сопровождающегося структурными изменениями. В ходе титрования не обнаружено сколько-нибудь значительных концентраций промежуточного соединения. Это свойство необычно для небольших молекул, и оно помогло Вильямсу и др. правильно установить строение витамина. Имеют ли эти реакции какое-либо биологическое значение? Тиольная форма, представленная выше, или «желтая форма» [уравнение (4-31)], могла бы присоединяться к активным центрам белков с помощью дисульфидных связей. Однако если такие реакции ферментов с тиамином и происходят, то пока они еще не обнаружены.
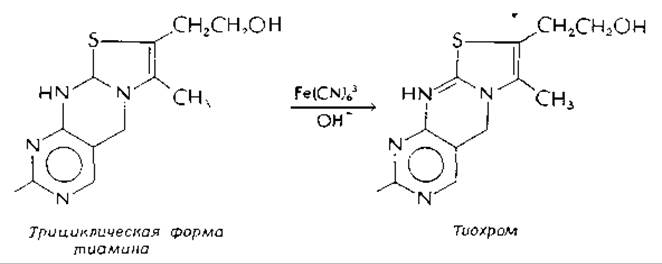
Щелочные условия ускоряют также деградацию тиамина. Таким образом, при приготовлении пищи в слабощелочных условиях тиамин разрушается Тиольная форма подвергается гидролизу и окислению в дисульфид кислородом воздуха. Трициклическая форма [уравнение (4-31)] окисляется в тиохром, флуоресцирующее соединение, образование которого из тиамина при обработке щелочным феррицианидом лежит в основе широко используемого флуориметрического метода определения тиамина:
Расщепление сульфитом. В растворе сульфита натрия при pH 5 тиамин расщепляется в результате реакции нуклеофильного замещения у метиленовой группы с образованием свободного тиазола и сульфокислоты. Аналогичное расщепление катализирует ферменты, расщепляющие тиамин (тиаминазы):

Аналоги. Замещение метальной группы у пиримидинового цикла на этильную, пропильную или изопропильную группы дает соединения с некоторой витаминной активностью, однако ее замещение водородом снижает активность до 5% от исходной. Бутильный аналог является конкурентным ингибитором. Обработка тиамина кипящей 5 н. НСl дезаминирует его в Оксианалог, окситиамин — мощный антагонист. Пиритиамин, другой конкурентный аналог, содержащий остаток вместо тиазолиевого цикла, очень токсичен, в особенности для нервной системы.

Суточная потребность: не менее 0,23 мг на каждые 1000 ккал потребляемой пищи за один день, но не менее 0,8 мг/день.