Биохимия - Химические реакции в живой клетке Том 2 - Д. Мецлер 1980
Типы реакций, катализируемых ферментами
Реакции нуклеофильного замещения (реакция типа 1)
Карбоний-ионы
Второй хорошо известный механизм нуклеофильного замещения состоит в том, что вначале отщепляется уходящая группа (часто в протонированной форме), оставляя карбоний-ион:
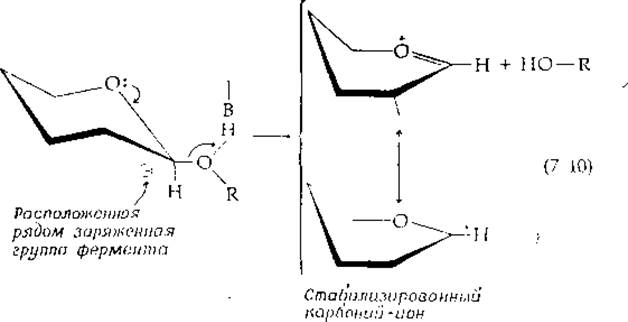
Согласно терминологии, принятой в физической органической химии, — это реакция типа SN1, а не реакция SN2-типa, показанная в уравнениях (7-3) и (7-6)1). Заметим, что карбоний-ион изображается как резонансный гибрид двух состояний, в одном из которых (оксоний-ион) имеется двойная связь между углеродом и кислородом. Можно себе представить, что такая структура с двойной связью образовалась из исходной структуры путем внутреннего замещения неподеленной парой электронов, принадлежащих кислороду, как это показано маленькими стрелками. Такой внутренний тип замещения можно рассматривать также как реакцию элиминирования. Следовательно, как это часто бывает при изучении ферментативных реакций, мы сталкиваемся с семантической проблемой. Является ли эта реакция элиминированием или только наполовину происшедшим замещением? В целях классификации метаболических реакций проще всего рассматривать процесс в целом как реакцию замещения.
В стабилизированном карбоний-ионе углеродные атомы цикла С-2, С-1 и С-5, а также атом кислорода находятся почти в копланарном положении. Такая конформация цикла называется полукреслом.
а. Лизоцим
Существует предположение, что механизм ферментативной реакции, включающий образование кабоний-иона, действует в случае лизоцима — фермента, основная роль которого состоит в атаке и расщеплении полисахаридных цепей пептидогликанового слоя клеточных стенок бактерий [10]. Лизоцим содержится в качестве защитного агента в слёзной жидкости, в других выделениях организма и в очень больших количествах в яичном белке. Лизоцим из яичного белка был первым ферментом, у которого методом дифракции рентгеновских лучей была определена полная трехмерная структура [11].
1) Эта терминология мало пригодна для ферментов, так как, хотя рассматриваемые реакции являются реакциями нуклеофильного замещения (Sn), распад фермент-субстратных комплексов с образованием продукта представляет собой обычно процесс нулевого порядка и цифры 1 и 2 в символах SN1 и SN2 обозначают порядок или молекуляриость реакции.
Оказалось, что топография шести колец N-ацетилглюкозамина или N-ацетилмурамовой кислоты в молекуле полисахаридного субстрата в точности соответствует впадине в молекуле лизоцима. При действии лизоцима связь между четвертым и пятым кольцами разрывается (рис. 2-9). В предполагаемом активном центре остаток глутаминовой кислоты (№ 35) находится в положении, точно соответствующем его роли донора протонов [т. е. ВН в уравнении (7-10)), тогда как остаток аспарагиновой кислоты (№ 52) лежит на противоположной стороне впадины. Как Glu-35, так и Asp-52 имеют аномально высокие значения рKа (микроскопические рKа составляют ∼5,3 и 4,6 соответственно)1) в полностью протонированном активном центре [12], что связано с гидрофобным окружением и наличием водородных связей с другими группами. Asp-52 обычно диссоциирует первым, и благодаря возникающему электростатическому взаимодействию Glu-35 остается протонированным вплоть до pH ~6. Расположенные рядом положительно заряженные основные группы влияют на величины рKа, и поведение фермента, следовательно, зависит от ионной силы среды [12]. Анион Asp-52 лежит близко (на расстоянии ~0,3 нм) к центру положительного заряда, ожидаемого в карбоннй-ионе [13], и, по-видимому, должен стабилизировать карбоний-ион [см. схему (7—10)].
Реакция, катализируемая лизоцимом, завершается стереоспецифическим присоединением гидроксильного иона к карбоний-иону. Продукт реакции сохраняет исходную ß-конфигурацию. Такая стереоспецифичность реакций ферментсодержащих карбоний-ионов не удивительна, поскольку фермент, вероятно, участвует в образовании и благоприятной ориентации атакующего гидроксильного иона2).
Как показывает изучение моделей, чтобы шесть сахарных колец субстрата были прочно связаны ферментом, кольцо, содержащее тот атом углерода, у которого происходит замещение, должно быть выведено из своего нормального состояния, соответствующего конформации «кресла», и перейти в форму «полукресла», необходимую для реализации механизма с участием карбоний-иона [15, 16]. Таким образом, в результате связывания полисахаридной цепи субстрата на шести различных центрах фермента происходит искажение конформации определенного цикла и возникает новая конформация, подобная конформации переходного состояния. Это, возможно, и является наиболее характерным аспектом ферментативного катализа.
1) При ионной силе ~0,2.
2) Механизм действия лизоцима четко описан в работе Дикерсона и Гейса [14].
б. Кинетические изотопные эффекты
Связь углерод—водород расщепляется легче, чем связь углерод — Дейтерий, и значительно легче, чем связь углерод — тритий. Таким образом, в тех случаях, когда предполагают, что стадия, лимитирующая скорость процесса, связана с разрывом связи С—Н, целесообразно сравнивать скорости расщепления связей С—44 и С—2Н. В случае реакции, катализируемой лизоцимом, медленная стадия не связана с разрывом связи углерод — водород, однако наблюдается вторичный кинетический изотопный эффект [17]. Различия в массе изотопов 1Н и 2Н приводят к небольшим различиям в колебательной энергии молекул, Содержащих этих изотопы. В результате молекула, содержащая 1Н в положении 1, может с несколько большей легкостью превращаться в карбоний-ион [уравнение (7-11)], чем молекула, имеющая 2Н в том же положении. Например, для неферментативного кислотного гидролиза фенилглюкозида, который, согласно имеющимся данным, протекает с
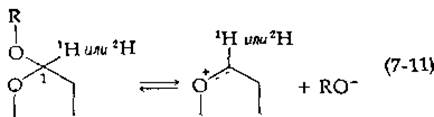
промежуточным образованием карбоний-иона, отношение k1H/k2H составляет 1,14. При гидролизе того же соединения, катализируемом основаниями (считают, что он протекает по механизму двойного замещения с участием соседней ОН-группы при С-2), отношение констант k1H/k2H равно 1,03. Отношение констант, измеренное для реакции, катализируемой лизоцимом, составляет 1,11, что гораздо ближе к соответствующему механизму с участием карбоний-иона, чем к механизму двойного замещения [17].
Как ни привлекательны аргументы, свидетельствующие о наличии в случае лизоцима механизма с участием карбоний-иона, все же имеющиеся экспериментальные данные можно объяснить наличием реакции двойного замещения, в процессе которой Asp-52 выступает в роли нуклеофила, участвующего в образовании лабильного промежуточного гликозилфермента. Интересно отметить, что как сахарозофосфорилаза, так и лизоцим содержат в активном центре карбоксилат-ион. В первом случае карбоксилат-ион образует содержащий ковалентную связь глюкозилфермент, в то время как в случае лизоцима карбоксилат-ион, видимо, стабилизирует карбоний-ион. Имеем ли мы дело действительно с различными механизмами? Не является ли образование глюкозилфермента артефактом, связанным с денатурацией сахарозофосфорилазы? Увы! Природа умеет хранить свои секреты. Трудности в выявлении тонких деталей механизма ферментативного действия заставляют исследователя проявлять известную долю скептицизма, рассматривать экспериментальные результаты и продумывать все возможные варианты, даже когда то или иное положение кажется бесспорным.