Принципы структурной организации белков - Г. Шульц 1982
Структурные основы механизма, действия и функции белков
Ферментативный катализ
Эффекты, способствующие высоким скоростям реакций
Сближенность, ориентация, направленность орбиталей и другие энтропийные эффекты
Ферменты могут ускорять химическую реакцию путем оптимальной для реакции ориентации реагентов в активном центре [741, 742]. Такая ориентация может включать и орбитальную направленность: точную взаимную ориентацию связывающих орбиталей атомов — партнеров по реакции [743, 744].
Связывание двух отдельных молекул в активном центре фермента превращает бимолекулярную реакцию в мономолекулярную, внутримолекулярную реакцию. Внутримолекулярные модельные реакции являются наиболее простым средством определения ускорения, которое может быть получено в результате сближения реагентов [631, 745]. Другими словами, энтропийный эффект фермента сказывается в увеличении эффективной концентрации субстрата. Поскольку скорости химических реакций пропорциональны концентрациям реагентов, то увеличения скорости в 103 раз можно ожидать на локальных участках с высокой концентрацией и упорядоченностью [631, 744].
Дестабилизация преобразуемых в субстратах атомных групп
Преобразуемая группа субстрата может быть дестабилизирована в тот момент, когда субстрат присоединяется к ферменту; энергия, затрачивающаяся на такую дестабилизацию [631], черпается из общей связывающей энергии. Механизмы дестабилизации могут включать замену растворителя, взаимодействия заряд — заряд и напряжения валентных связей и валентных углов. Дестабилизация может быть ослаблена в переходном состоянии, что означает снижение энергии активации, необходимой для достижения переходного состояния.
Помещая химические группы в неполярное окружение, можно увеличить скорость в 50 000 раз. Простой механизм, предложенный для пируватдекарбоксилазы, — тиаминпирофосфатсодержащего фермента основан на двух наблюдениях [746]. Одно состоит в том, что аддукт пирувата и аналог кофактора (рис. 11.3) декарбоксилируется в органических растворителях в 104 : 105 раз быстрее, чем в воде; второе относится к гидрофобности пируватсвязывающего центра в пируватдекарбоксилазе. Увеличение скорости декарбоксилирования, ожидаемое при переходе аддукта пируват — кофермент в такое окружение, вносит существенный вклад в наблюдаемую скорость ферментативного действия. Связывающую энергию, необходимую для фиксации заряженного субстрата в таком невыгодном окружении и для обеспечения увеличения скорости реакции, могут предоставить пирофосфатная группа и пиримидиновый цикл кофермента.
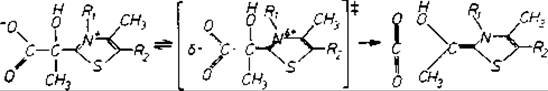
Рис. 11.3. Катализ декарбоксилирования при замене растворителя. Реакционноспособным фрагментом тиаминпирофосфата, кофактора ферментов декарбоксилирования, является тиазольная группа. В тиаминпирофосфате R1 — пиримидиновый, а R2 — пирофосфатный фрагменты; в обсуждаемом модельном соединении R1 = CH3 н R2 = Н. В органических растворителях модельное соединение теряет СO2 в 50 000 раз быстрее, чем в воде [746]. Предполагается, что органические растворители благоприятствуют переходному состоянию, показанному на рисунке в скобках.
Фрагменты ADP коферментов, таких, как ATP, FAD, NAD(P) или кофермент А, могут выполнять функцию, аналогичную функции пиримидиновой и пирофосфатной групп тиаминпирофосфата. Во всех изученных случаях (разд. 10.4) кофакторы присоединяются к своим коферментам в вытянутых конформациях, обеспечивая тем самым большое число взаимодействий с ферментами (рис. 11.4). Связывающая энергия, которая расходуется на специфические нужды других фрагментов кофермента, при этом всегда предоставляется фрагментом ADP.
Заряженные группы фермента становятся более активными, если сольватирующие молекулы воды заменить субстратом. Дестабилизация, основанная на десольватации, по-видимому, вносит вклад в каталитическое действие иона Zn2+, связанного с карбоксипептидазой А [747]. Замещение сольватирующей молекулы воды на субстрат уменьшает диэлектрическую проницаемость окружения иона металла и увеличивает его активность в поляризации ацильной группы субстрата, необходимой для нуклеофильной атаки. Аналогичный эффект вносит вклад в процессы в D-субцентре лизоцима. Взаимодействие с субстратом десольватированной карбоксильной группы остатка Asp-42 ведет к локальной дестабилизации фермент-субстратного комплекса; эта дестабилизация ослабляется при образовании переходного состояния, напоминающего оксокарбониевый ион [531].
Длины связей и валентные углы в субстрате могут искажаться при его присоединении к ферменту. Фермент может действовать на химическую группу субстрата, придавая ей строение, близкое к структуре переходного состояния; механизм такой структурной дестабилизации включает деформацию валентных углов, сближение реагирующих атомов до межатомных расстояний, меньших суммы вандерваальсовых радиусов, а также растяжение ковалентных связей до длины, превосходящей сумму ковалентных радиусов связанных атомов [631, 739, 740]. Наиболее хорошо изучен случай структурной дестабилизации, происходящей при присоединении N-ацетиламино-сахарного остатка, к субцентру D-лизоцима [748]. Тетраэдрический (sp)3-aтом углерода-1 искажен, и соответствующее напряжение ослабляется в переходном состоянии, где атом углерода-1 становится планарным (sp2). Если учитывать десольватацию остатка Asp-42 при связывании субстрата, то энергия дестабилизации составит по крайней мере + 8,6 ккал/моль. Эта энергия восполняется общей связывающей энергией олигосахаридного субстрата.
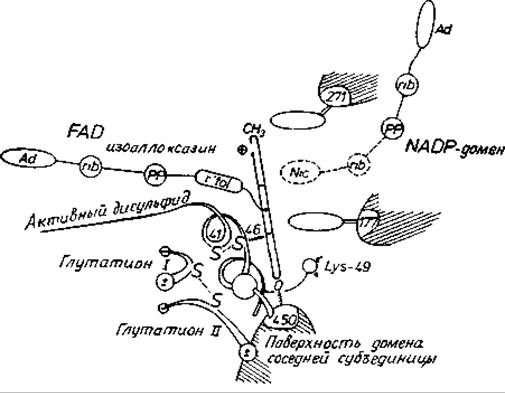
Рис. 11.4. Активный центр глутатнонредуктазы [124].
С димерном ферменте имеются два идентичных активных центра. Восстанавливаемые агенты проходят следующий путь (справа налево): никотинамидный фрагмент NADPH→изоаллоксазиновый цикл FAD (показан сбоку)→дисульфид с окислительно-восстановительной активностью→дисульфидная связь глутатиона. Глутатионсвязывающий центр и каталитический центр образованы остатками обеих субъединиц. Отметим, что NADPH и FAD присоединяются к ферменту в вытянутых конформациях.
Связывающие взаимодействия вдали от каталитического центра стабилизируют комплекс трипсин — ингибитор. Это следует из сравнения взаимодействия трипсин — субстрат с взаимодействием трипсин — ингибитор [269, 5361. Комплекс трипсина с псевдосубстратом — ингибитором характеризуется аномальным расстоянием
2,6 А между атомом углерода карбонильной группы-15 псевдосубстрата и остатком Sеr-195фермента. Обычным субстратам химотрипсина и трипсина, в которых осуществляется несколько выгодных контактов, для достижения стадии ацилирования необходима энергия активации от +5 до +15 ккал/моль. Однако при образовании комплекса трипсин — ингибитор оптимизируются многочисленные другие взаимодействия, и величина ΔG оказывается равной —18 ккал/моль, несмотря на напряженность С—Оy -аддукта (табл. 5.6). Таким образом, различие энергий стабилизации можно объяснить различием контактирующих областей в комплексах, которые трипсин образует с ингибитором и с обычными субстратами [749].
Процессы в каталитическом центре могут стабилизировать переходное состояние. До сих пор подчеркивался тот факт, что дальние взаимодействия поставляют свободную энергию активируемым группам в каталитическом центре фермент-субстратного комплекса. Однако взаимодействия и в самом каталитическом центре могут стабилизировать переходное состояние и тем самым вносить вклад в эффективность ферментативного катализа. В химотрипсине выигрыш энергии, обеспечивающийся образованием двух водородных связей между активированным субстратом и атомами азота остова, а также частичной компенсацией заряда скрытого внутри белка остатка Asp-102 (рис. 11.1), способствует компенсации энергии образования напряженной связи между ферментом и субстратом в тетраэдрическом комплексе [537].
Химические группы как части ферментов
Дестабилизирующие эффекты в фермент-субстратном комплексе оказывают влияние на состояние преобразуемых групп субстратов. Однако в ферменте предусмотрены также функциональные группы, которые более тонко воздействуют на преобразуемые группы. Общий кислотно-основной катализ довольно обычен в ферментах, и с его помощью скорость реакции может увеличиваться в 1000 раз. В химотрипсине эту функцию выполняет зарядно-релейная система, которая посредством водородных связей обеспечивает протонный транспорт в нескольких стадиях реакции (рис. 11.1). В других ферментах, например в глутатионредуктазе, белок обладает активными группами (FAD и цистеиновая пара с окислительно-восстановительной активностью) для транспорта электронов через молекулу фермента (рис. 11.4).
Раздельное рассмотрение эффектов, приводящих к увеличению скорости ферментативного катализа
Неоднократно предпринимались попытки количественно оценить вклады различных эффектов (например, сближенности, направленности орбиталей, дестабилизации, общего кислотно-основного катализа и др.) в увеличение скорости, к которому приводит действие данного фермента. Однако пример химотрипсина показывает, что эти «эффекты» представляют разные способы описания одного и того же действия в активном центре и в действительности их нельзя отделить друг от друга. С другой стороны, представления, развитые при анализе фермент-субстратных взаимодействий, расширили наши представления о химическом катализе и способствовали созданию аналогов ферментов на основе полимеров непептидной природы [745, 750].