ОСНОВИ МЕДИЧНОЇ БІОЛОГІЇ - 2012
Популяційно-статистичний метод. Медико-генетичне консультування
Популяційно-статистичний (популяційно-генетичний) метод дозволяє вивчати: генетичний склад популяцій людей, частоту нормальних і патологічних генів і генотипів у різних популяціях: гетерозигот, гомозигот домінантних і рецесивних, частоту нормальних і патологічних фенотипів. Частота певного генотипу (алеля) - це відносне число особин у популяції з даним генотипом (алелем). Частоту виражають у відсотках або частках одиниці. Наприклад, частота блакитнооких людей у популяції - 49%, або 0,49. Метод ґрунтується на спостереженні спадкових ознак у великих групах населення. Проводяться безпосереднє вибіркове дослідження частини популяції або вивчаються архіви лікарень та інших медичних закладів, а також проводиться опитування населення шляхом анкетування. Вибір способу залежить від мети дослідження. Останній етап полягає у математичному аналізі одержаних даних.
Одним із найбільш простих і універсальних методів аналізу частот генотипів та генів в популяції є метод запропонований Г. Харді і В. Вайнбером. У 1908 році математик Г. Харді в Англії і лікар-антрополог В. Вайнберг у Німеччині сформулювали закон підтримки генетичної
рівноваги в ідеальній популяції. Ними було запропоновано для відображення розподілу генотипів у панміктичній популяції застосувати формулу бінома Ньютона: (a + b) = a2 + 2ab + b2.
Частота генотипів і фенотипів розраховується за формулою Харді-Вайнберга:
p2 + 2pq + q = (p + q) = 1(або 100 %),
де p - частота домінантного гена (А); q - частота рецесивного гена (а); p - частота гомозигот за домінантним геном (АА); q - частота гомозигот за рецесивним геном(аа); 2pq - частота гетерозигот (Аа).
За цим рівнянням можна визначити генетичну структуру популяції без проведення спеціальних досліджень. Для цього треба знати лише частоту рецесивних гомозигот (аа) - q . Це єдиний генотип, який можна визначити за фенотипом.
Приклад. У популяції частота резус-від'ємних людей (аа) складає 15%. Визначити частоту резус-позитивних гомозигот (АА) і частоту резус-позитивних гетерозигот (Аа) в популяції.
Розв'язання. Частота генотипу аа (q2) = 15 % = 0,15. Звідси частота алеля a(q) = √0,15 = 0,387. Частота гена A(p) = 1 — q = 1 - 0,387 = 0,613. Частота генотипу АА(р2) = 0,6132 = 0,376. Частота генотипу Aa(2pq) — 2 x 0,613 x 0,387 = 0,474. Отже, резус-від'ємних особин у популяції - 15 %, резус-позитивних - 85 % (серед них гомозигот - 37,6 %, гетерозигот - 47,4 %).
Розроблено ряд інших спеціальних математичних методів аналізу генетичної структури популяцій та процесів, які відбуваються на цьому рівні життя. Досліджувані популяції розрізняються за біологічними ознаками, географічними умовами життя, економічним станом. Вивчення поширення генів на певних територіях свідчить про те, що їх можна розділити на такі категорії:
1) мають універсальну поширеність, такі гени зустрічаються у всіх регіонах і популяціях, але з різною частотою (до них відноситься більшість відомих генів - це гени фенілкетонурії, галактоземії, гемофілії, деяких форм розумової відсталості). Наприклад, ген фенілкетонурії зустрічається з частотою 1 % у населення Європи; ген дальтонізму з частотою 7 % у чоловіків та 13 % - у жінок (у гетерозиготному стані).
2) зустрічаються локально, переважно у певних регіонах, популяціях; наприклад, ген серпоподібно-клітинної анемії, який поширений у країнах Африки і Середземномор'я, де розповсюджена малярія.
Популяційно-статистичний метод застосовують для вивчення:
1) частоти генів у популяціях, включаючи частоту спадкових хвороб;
2) мутаційного процесу;
3) ролі спадковості й середовища у виникненні хвороб, особливо хвороб із спадковою схильністю;
4) ролі спадковості й середовища у формуванні фенотипового поліморфізму людини за нормальними ознаками;
5) значення генетичних чинників в антропогенезі, зокрема в расоутворенні (Н.П. Бочков, 2001).
Знання генетичного складу популяцій населення має велике значення для медицини, зокрема, для соціальної гігієни, медичної генетики, медико-генетичного консультування.
Медико-генетичні аспекти сім'ї
У людини алельний склад генотипів нащадків залежить від системи шлюбів. Розрізняють невибіркові і вибіркові шлюби, аутбридинг, інбридинг, інцестні шлюби. При невибіркових шлюбах (панміксії) існує однакова ймовірність вступу в шлюб будь-якого індивідуума з будь-яким індивідом протилежної статі. Вибіркові (асортативні) шлюби - це шлюби між людьми зі схожими фенотипами. Розрізняють позитивні і від'ємні асортативні шлюби. У першому випадку має місце більш частіший, а в другому - більш рідкіший вступ в шлюб індивідуумів, схожих за фенотипом. Так, шлюби між глухонімими, а також між людьми, схожими за ростом або розумовими здібностями, спостерігаються частіше, ніж можна було б чекати при панміксії. Навпаки, рудоволосі чоловіки і жінки уникають втупати в шлюби один з одним. Аутбридинг - неспоріднені шлюби, інбридинг - споріднені шлюби. Інцестні (заборонені) шлюби - форма інбридингу, шлюби між родичами першого ступеню спорідненості (батько - дочка, брат - сестра). В історії людства така форма інбридингу як система існувала в династії Птоломеїв, які правили в Єгипті в 305-30 pp. до н.е. У даний час такі шлюби заборонені майже повсюдно. У багатьох державах заборонені шлюби племінниця (племінник) - дядя (тітка). У США більше як у третині штатів заборонені шлюби між двоюрідними братами і сестрами. У високогірських селах Швейцарії, єврейських общинах багатьох міст Германії частота шлюбів між двоюрідними родичами складає 6-12%. Висока частота споріднених шлюбів є типовою для малих за розмірами груп людей, ізольованих через географічні, економічні, релігійні, національні і расові особливості.
Медико-генетичне значення різних систем шлюбів: 1) інбридинг збільшує гомозиготність, у тому числі за патологічними генами; у близькоспоріднених шлюбах збільшується дитяча смертність; збільшується ймовірність появи спадкових хвороб з автосомно-рецесивним типом успадкування; 2) аутбридинг підтримує високий рівень гетерозиготності, що може обумовити підвищення життєздатності.
Медико-генетичні аспекти сім'ї. Останніми роками істотно змінилася структура захворюваності населення. Природжені вади розвитку і спадкові захворювання стали переважати, а в структурі дитячої смертності займають перше місце. Близько 5 % дітей народжуються з різними генетичними дефектами. В ідеалі кожна сімейна пара повинна пройти повне медико-генетичне обстеження до зачаття, лише тоді планувати народження дітей. При цьому за допомогою лікаря формується правильне ставлення до контрацепції, вагітності, сімейного життя, догляду за дитиною. На сучасному етапі можливе запліднення поза організмом in vitro з наступною імплантацією зародка в матку, що збільшує ймовірність репродукції.
Медико-генетичне консультування
Медико-генетичне консультування (МГК) - один з видів спеціалізованої допомоги населенню, спрямований головним чином на попередження появи в сім'ї хворих зі спадковою та природженою патологією. Медико-генетичне консультування свої витоки бере з 20-х років ХХ сторіччя і пов’язане з ім’ям російського генетика і невропатолога С.М. Давиденкова. Пізніше, у 40-х роках американський учений Д. Нель організував перший кабінет із медико-генетичного консультування.
Основні завдання МГК: 1) встановлення точного діагнозу спадкової хвороби; 2) встановлення типу успадкування захворювання в даній сім'ї; 3) розрахунок ризику повторення спадкової хвороби в сім'ї; 4) визначення найбільш ефективного способу профілактики; 5) пояснення тим, хто звернувся за допомогою, змісту зібраної інформації, медико-генетичного прогнозу і методів профілактики.
МГК проводиться в чотири етапи: 1) встановлення діагнозу; 2) складання прогнозу; 3) висновки або заключення; 4) порада сім'ї щодо профілактики народження хворої дитини.
На першому етапі проводиться уточнення клінічного діагнозу за допомогою генетичних методів. На другому - складається прогноз імовірності народження хворої дитини (генетичний ризик). Складання прогнозу залежить від типу успадкування. Для менделюючих (моногенних) хвороб оцінка генетичного ризику проводиться шляхом розрахунків на основі законів Г. Менделя; для полігенних хвороб — переважно на основі емпіричних даних, тобто на фактичних спостереженнях. На третьому - лікар-генетик дає заключення з рекомендаціями щодо ступеня генетичного ризику. Генетичний ризик до 5 % розцінюється як низький і не вважається протипоказанням до дітонародження в даній сім'ї. Ризик від 6 до 20 % вважається середнім. У цьому випадку рекомендації залежать від величини генетичного ризику, а також від медичних і соціальних наслідків даної патології, можливості пренатальної діагностики. Якщо генетичний ризик перевищує 20 %, то при відсутності методів пренатальної діагностики даної патології подальше народження дітей не рекомендується. На четвертому - лікар-генетик дає поради щодо профілактики народження хворої дитини.
Принципово важливе завдання МГК - знайти прості і доступні методи виявлення гетерозиготних носіїв мутантного гена, провести диференційну діагностику з фенокопіями, новими мутаціями. Практично кожна сімейна пара повинна пройти МГК до планування народження дітей (проспективне) і обов'язково після народження хворої дитини (ретроспективне). Обов'язкове МГК повинно проводитись у сім'ях, що належать до групи ризику за такими показаннями:
1) народження дитини з природженою вадою розвитку;
2) встановлена спадкова хвороба або підозра на неї у сім'ї:
а) один з батьків є носієм збалансованої транслокації хромосом;
б) мати є носієм патологічного гена, зчепленого з Х-хромосомою;
в) діти мають природжені вади розвитку або хвороби обміну речовин;
3) затримка фізичного розвитку або розумова відсталість у дитини;
4) повторні спонтанні аборти, викидні, мертвонароджені не з’ясованої етіології;
5) близькоспоріднені шлюби;
6) вплив тератогенів або підозрілих на тератогенність факторів у перші три місяці вагітності;
7) несприятливий перебіг вагітності;
8) вік вагітної понад 35 років;
9) підозра при УЗД на природжену ваду розвитку плода.
Для надання медико-генетичного консультування в Україні існує структурний підрозділ у системі охорони здоров'я (при поліклініках, лікарнях) - медико-генетична служба. До її складу входять: 53 міжрайонних медико-генетичних кабінети (ММГК) і один міський, 18 обласних медико-генетичних консультацій (ОМГК), 7 міжобласних медико-генетичних центрів - ММГЦ (Київський, Львівський, Харківський, Криворізький, Кримський, Донецький, Одеський). У 1992 р. створений Український науковий центр медичної генетики МОЗ і Академії медичних наук України. Йому і Львівському науково-дослідному інституту спадкової патології доручено координацію всіх розділів роботи медико-генетичної служби України. Керує роботою всієї медико-генетичної служби Координаційна рада з медичної генетики при Міністерстві охорони здоров'я (МОЗ) України і головний спеціаліст МОЗ України. Спеціалізовану допомогу хворим із спадковими хворобами надають також спеціалісти профільних науково-дослідних інститутів МОЗ України, кафедр медичних вищих наукових закладів та інститутів удосконалення лікарів. Всі методи медико-генетичної допомоги регламентуються наказом МОЗ України № 203 від 18.10.1988 «Про заходи з поліпшення медико-генетичної допомоги в Україні» та наказом МОЗ України № 503 від 28.12.2002 «Про удосконалення амбулаторної допомоги населенню України».
Пренатальна діагностика спадкової патології
Пренатальна (допологова) діагностика дає можливість діагностувати природжені вади розвитку або спадкові захворювання плода на ранніх стадіях його розвитку. Рання діагностика допомагає або прийняти рішення щодо переривання вагітності, або підготувати родину до народження хворої дитини. Пренатальна діагностика призначається в таких випадках:
1. Вік матері визначений у 35 років, батька - старше 40 років.
2. Наявність у родині попередньої дитини з хромосомною патологією, у тому числі із синдромом Дауна (попередній анеусомік).
3. Поява в сім’ї точно встановленого спадкового захворювання.
4. Перебудови батьківських хромосом.
5. Наявність у родині захворювань, успадкованих зчеплено зі статтю.
6. Поява у матері Х-зчепленого рецесивного патологічного гена;
7. Синдром фрагільної Х-хромосоми.
8. Гемоглобінопатії.
9. Природжені помилки метаболізму.
10. Різні спадкові захворювання, що діагностуються методом зчеплення з ДНК-маркерами.
11. Дефекти нервової трубки невстановленої етіології, діти з множинними уродженими вадами розвитку і з хромосомною патологією.
12. Поява структурних перебудов хромосом (особливо транслокацій та інверсій) в одного з батьків.
13. Гетерозиготність обох батьків по одній парі алелів при автосомно-рецесивних захворюваннях.
14. Вагітні із зони підвищеного радіаційного забруднення, із тератогенним впливом та ін.
15. Спостерігається багатоводдя або маловоддя під час даної вагітності.
16. Підвищення рівня альфа-фетопротеїну в крові більше 2,5 МоМ або зниження до 0,5 МоМ.
17. Підозра при УЗС на природжену ваду розвитку плода.
18. Були вірусні захворювання в І триместрі вагітності.
19. Екстрагенітальні захворювання вагітної (гіпертонічна хвороба, цукровий діабет, захворювання щитоподібної залози, природжені та набуті вади серця).
Методи допологової діагностики можна розділити на три групи: 1) просіюючі; 2) неінвазивні і 3) інвазивні.
Просіюючі методи. Вони дозволяють виділити жінок, які мають підвищений ризик народження дитини із спадковою або природженою патологією. До цих методів відносяться лабораторні методи визначення рівнів а (альфа)-фетопротеїну, хоріонічного гонадотропіну, некон'югованого естріолу. Альфа-фетопротеїн (а-ФП) - білок плодового походження. Він продукується спочатку жовтковим мішком, а з кінця І триместру - печінкою плода. Його вміст змінюється протягом вагітності, а також залежить від екологічних умов. Визначення вмісту а-ФП проводять у сироватці крові матері та в амніотичній рідині в період 16-18 тижнів вагітності. Підвищення рівня а-ФП може свідчити про вади розвитку нервової трубки, черевної стінки, інших систем. Зниження рівня а-ФП спостерігається при хромосомній патології (хвороба Дауна, синдром Едвардса), смерті плода. Доволі ефективним методом для діагностики синдрому Дауна є визначення рівня хоріонічного гонадотропіну в сироватці матері. Після першого триместру вагітності він повинен знижуватись. Коли мати виношує плід з хромосомною патологією, концентрація даного гормону залишається незмінною. При синдромі Дауна значно нижчим є рівень некон'югованого естріолу в сироватці крові вагітної.
Неінвазивні методи - це методи, які не потребують оперативного втручання. Найбільш часто використовується ультразвукове дослідження.
Ультразвукове дослідження (УЗД, ехографія, ультрасонографія). Метод заснований на пропусканні через черевну порожнину ультразвукових хвиль, які після відображення від поверхні тканин плода вловлюються і посилюються, що дозволяє отримати зображення обрисів плаценти і органів плода. Джерелом і приймачем ультразвукових хвиль є датчик. При неускладненому перебігу вагітності УЗД проводиться два рази: на 20-24 і 28-32 тижні. Під час УЗД виявляють вади розвитку плода, патологію з боку центральної нервової системи: гідроцефалія (водянка головного мозку), мікроцефалія (малий розмір головного мозку), аненцефалія (відсутність головного мозку); шлунково-кишкового тракту, нирок: агенезія (відсутність) або гіпоплазія (недорозвинення), гідронефроз, полікістоз; органів дихання, серцево-судинної системи, аномалії скелета, пухлини плода та ін. Існують застереження щодо застосування УЗД у ранні строки вагітності, оскільки ніхто не довів як шкідливого, так і нешкідливого впливу ультразвуку на організм плода.
Інвазивні методи. До інвазивних методів пренатальної діагностики спадкових хвороб відносять: біопсію ворсин хоріона, амніоцентез, кордоцентез, біопсію тканин плода, фетоскопію.
Біопсія ворсин хоріона і дослідження трофобласта проводиться в першому триместрі вагітності (9-11-й тиждень) під контролем УЗД. Процедура здійснюється трансабдомінальним або трансцервікальним шляхом. Отримані клітини хоріона досліджують для визначення хромосомного набору, ДНК, ферментів, статі плода, наявності гемоглобінопатій (серпоподібноклітинна анемія, Р-таласемія).
Амніоцентез — вилучення навколоплодових вод за допомогою тонкої голки для отримання і дослідження клітин плода, які в них знаходяться (рис.). Найпоширенішим є трансабдомінальний метод (через стінку черевної порожнини). Проводиться під ультразвуковим контролем у період 12-18 тижнів вагітності. Клітини плода досліджують або відразу, або культивують протягом 2-4 тижнів для наступних цитогенетичних, молекулярно- генетичних, біохімічних досліджень. Метод дає можливість виявити хромосомні аномалії, порушення обміну речовин.
Кордоцентез - пункція судин пуповини плода для забирання крові. Проводиться під ультразвуковим контролем. Зразки крові використовуються для таких же цілей, як і при амніоцентезі. Однак кордоцентез має ту перевагу, що кров є більш зручним об'єктом для досліджень, ніж клітини амніотичної рідини. Клітини крові культивуються швидше (протягом 2-3 днів) і надійніше, ніж амніоцити.
Біопсія тканин плода здійснюється в 2-му триместрі вагітності під контролем УЗД без фетоскопії. Для діагностики спадкових хвороб шкіри (іхтіоз, бульозний епідермоліз) роблять біопсію шкіри. Матеріал досліджують під світловим або електронним мікроскопом. Для діагностики м'язової дистрофії Дюшена роблять біопсію м'язів з наступним дослідженням імунофлуоресцентним методом, який виявляє білок дистрофін. У хворих такий білок відсутній, оскільки не функціонує нормальний ген, який його виробляє.
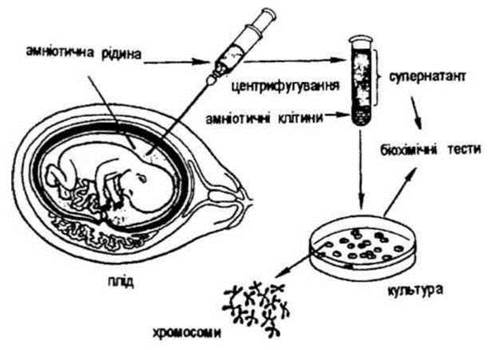
Рис. 36. Амніоцентез - метод пренатальної діагностики спадкових хвороб.
Фетоскопія - візуальне дослідження плода за допомогою ендоскопа (фетоскопа). Ендоскоп, введений в амніотичну порожнину (через передню черевну стінку або заднє склепіння) дає можливість оглянути частини плода, провести забір крові, біопсію шкіри для аналізу. Метод дозволяє діагностувати видимі природжені вади (полідактилія, ахондроплазія тощо), виявити при дослідженні біоптатів шкіри іхтіоз, бульозний епідермоліз. За цим методом також можна діагностувати спадкові гемоглобінопатії, еритроцитарні ензимопатії, імунодефіцитні стани. Проводиться лише у випадках особливих показань на 18-23-му тижнях вагітності під контролем УЗД. У 7-8% випадків після фетоскопії зареєстровані викидні.
Останніми роками розвивається гіпотеза про преконцептивну профілактику. Вона починається за кілька місяців до зачаття і продовжується протягом ранніх строків розвитку зародка. Підготовка організму матері (вітамінізація, антиоксидантна терапія, підвищення імунітету, відсутність стресів) до зачаття і дотримання даних умов на ранніх строках розвитку зародка (до 10 тижнів) сприяє зменшенню частоти природжених вад розвитку мультифакторної природи. За даними М.П. Бочкова (1997), у жінок, яким проводилась додаткова вітамінізація, частотність повторного народження дитини з вадами нервової трубки зменшувалась від 4,6 до 0,7 %.