ОСНОВИ МЕДИЧНОЇ БІОЛОГІЇ - 2012
Молекулярні хвороби. Біохімічний метод та ДНК-діагностика Генні (молекулярні) хвороби
Генними називаються спадкові хвороби, які зумовлені генними мутаціями. Причини - порушення хімічної структури гена (молекули ДНК). Тому генні хвороби називають молекулярними. Класичні генні мутації успадковуються як менделюючі ознаки. Вони зумовлені мутацією одного гена (моногенні хвороби). Генеративні мутації розглядаються як повні форми. Відомі також мозаїчні форми, коли мутації виникають на ранніх стадіях дроблення зиготи в одній клітині. Такий організм буде мозаїчним за даним геном: частина клітин міститиме нормальний алель, частина - мутантний. Діагностувати мозаїчні форми генних хвороб можна сучасними молекулярно-генетичними методами.
Розрізняють моногенні і полігенні хвороби. Моногенні хвороби зумовлені дією одного мутантного гена. Полігенні хвороби визначаються множинними генами, прояв яких залежить від факторів зовнішнього середовища. Моногенні хвороби успадковуються згідно із законами Г. Менделя. Залежно від типу успадкування розрізняють такі моногенні хвороби - автосомно- домінантні, автосомно-рецесивні, Х-зчеплені домінантні, Х-зчеплені рецесивні, Y-зчеплені.
Аутосомно-домінантні хвороби зумовлені мутацією домінантного гена в аутосомі. Патологічна ознака зустрічається в кожному поколінні. Ген фенотипово проявляється як у гомозиготному АА, так і гетерозиготному Аа станах за умови повної пенетрантності та експресивності. У гомозигот перебіг хвороби більш тяжчий, ніж у гетерозигот. Рецесивні гомозиготи аа не хворіють. До аутосомно-домінантних хвороб належать: синдром Марфана, ахондроплазія, хорея Гентингтона, поліпоз нирок, аніридія (відсутність райдужної оболонки), гіперхолестеринемія, хвороба Штейнерта, полідактилія (6 і більше пальців), брахідактилія (короткопалість).
Аутсомно-рецесивні хвороби зумовлені мутацією рецесивного гена в аутосомі. Тип успадкування - аутосомно-рецесивний. Хворіють лише рецесивні гомозиготи (аа), гетерозиготи (Аа) не хворіють, але є носіями патологічного гена. До цієї групи спадкових хвороб належать: фенілкетонурія, галактоземія, муковісцидоз, спінальна м'язова атрофія, первинний гемохроматоз, амавротична ідіотія та ін.
Х-зчеплені домінантні хвороби зумовлені мутацією домінантного гена в Х-хромосомі. Особливість - у хворого батька (XAY) всі дочки будуть хворі, а всі сини - здорові. До цієї групи хвороб належать: гіпофосфатемія (вітамін D-резистентний рахіт), синдром Гольтца (дермальна гіпоплазія фокальна), синдром Коффіна-Лоурі (розумова відсталість і кістково- хрящові аномалії).
Вітамін D-резистентний рахіт (гіпофосфатемія) - рахіт, який не піддається лікуванню вітаміном D. Гіпофосфатемію можна виявити відразу після народження, а ознаки рахіту з'являються в кінці першого - на початку другого року життя, коли діти починають ходити. Найбільш вираженими є зміни нижніх кінцівок-викривлення довгих трубчастих кісток. Характерні низький зріст, обмеження рухомості у великих суглобах, доліхоцефалія, дисплазія (порушення формування) нігтів. Захворювання зумовлене зниженням реабсорбції фосфатів в канальцях нирок.
Х-зчеплені рецесивні хвороби зумовлені мутацією рецесивного гена в Х-хромосомі. Тип успадкування - Х-зчеплений рецесивний. До цієї групи хвороб належать: гемофілія, дальтонізм, м'язова дистрофія Дюшена, синдром Леша-Ніхана, ін.
Гемофілія є класичним прикладом Х-зчепленого рецесивного аномального гена, який проявляється фенотипово в чоловіків. У жінок-гетерозигот його дія пригнічується домінантним алелем нормального зсідання крові. Батьки-гемофіліки ніколи не передають ген гемофілії синам. Тому їхні діти здорові, але всі дочки народжуються носіями хвороби. Розрізняють різні клінічні прояви гемофілії - від легких кровотеч до масових крововиливів. Імовірно, це залежить від різних мутацій одного гена. Найбільш поширені дві форми гемофілії - А і В. Вони характеризуються відсутністю в плазмі крові різних антигемофільних глобулінів. Частотне співвідношення гемофілії А і В становить 5:1. Обидві форми гемофілії зустрічаються з частотою 1:5000 новонароджених хлопчиків.
Дальтонізм є однією з найпоширеніших спадкових хвороб, що успадковуються з X-хромосомою. Характеризується порушенням сприймання кольорів (переважно червоного і зеленого). Принципи його успадкування такі самі, як гемофілії.
Y-зчеплені хвороби. Мутантний ген локалізований у негомологічній ділянці Y-хромосоми. Успадкування - виключно по чоловічій лінії. Приклади - гіпертрихоз, іхтіоз.
Моногенні хвороби також поділяються на групи залежно від характеру порушень:
- ензимопатії (ферментації);
- дефекти структурних і транспортних білків;
- порушення циркулюючих білків крові;
- генні хвороби з невідомим первинним біохімічним дефектом.
Відповідно класифікації ВООЗ, моногенні або молекулярні хвороби поділяються на такі групи, які визначаються порушенням:
1) амінокислотного обміну: фенілкетонурія, тирозинемія, алькаптонурія, гомоцистинурія, цистинурія, хвороба Хартнупа, триптофанемія, хвороба «кленового сиропу», гістидинурія, гістидинемія та інш.;
2) вуглеводного обміну: галактоземія, фруктоземія, глікогенози, синдром мальабсорбції вуглеводів, мукополісахаридози;
3) ліпідного обміну: гіперліпопротеїдемії, сфінголіпідози (хвороба Німанна-Піка), гангліозидози (хвороба Тея-Сакса);
4) стероїдного обміну: адреногенітальний синдром;
5) пуринового та піримідинового обмінів: синдрои Леша-Найана;
6) обміну речовин у сполучній тканині, кістках та м’язах: Хвороба Марфана;
7) структури гема та порфірина: гемаглобінопатії, серпоподібноклітинна анемія, таласемія;
8) обміну речовин в еритроцитах та порушення їх структури: спадковий мікросфероцитоз;
9) аномалій обміну металів;
10) хвороби, які характеризуються дефектом транспорту різних речовин;
11) хвороби, які характеризуються аномаліями структури: функції ферментів та білків плазми.
Основою генних хвороб є мутації відповідних генів, які призводять до синтезу ферментів зі зміненою активністю. У гомозигот активність ферменту низька, що й призводить до порушення конкретного обміну, у гетерозигот активність часто складає ≈ 50%, тому хвороби здебільшого успадковуються за А-Р або Х-Р типом.
Метаболічні зміни відбуваються за схемою:
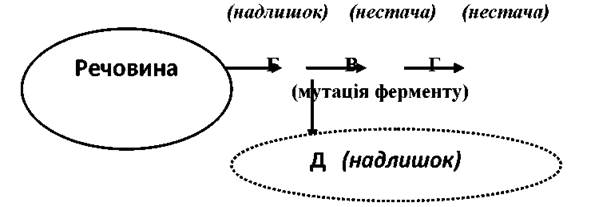
Тому, при мутації ферменту відбувається надлишок одних речовин (Б, Д) та нестача інших продуктів (В, Г). Ферментопатії (ензимопатії) складають найбільш численну групу генних хвороб. Фермент або змінює свою структуру і функціональні властивості, або зовсім не утворюється. У такому випадку, біохімічна реакція, що відбувається за участю даного фермента, блокується. При цьому може мати місце: 1) недостатнє утворення продуктів даної реакції або більш віддалених продуктів його перетворення; 2) нагромадження в організмі субстрату блокованої реакції або його попередників; 3) зміна основного напряму в перебігу реакції і підвищене утворення продуктів, які в нормі є в незначних кількостях.
Діагностика ензимопатій здійснюється за допомогою біохімічних методів. Рання діагностика та медикаментозна чи дієтична корекція дають можливість лікувати та запобігти розвитку цих спадкових хвороб.
Фепілкетонурія — спадкова хвороба - ферментопатія, зумовлена генетичним дефіцитом фермента фенілаланінгідроксилази, який необхідний для перетворення амінокислоти фенілаланіну в тирозин. Це веде до нагромадження фенілаланіну, фенілпірувату та фенілацетату у крові, спинномозковій рідині, тканинах і їх токсичної дії на ЦНС. Діти народжуються здоровими, але з надходженням в організм фенілаланіну з молоком матері поступово розвивається розумова відсталість. Оскільки порушення обміну фенілаланіну веде до зниження рівня тирозину, у хворих спостерігається зменшення пігментації шкіри, волосся, райдужної оболонки. Частота фенілкетонурії в європейських популяціях складає в середньому 1:10000 новонароджених. Тип успадкування автосомно-рецесивний. Ген фенілаланінгідроксилази розташований у хромосомі 12.
Клінічні діагностичні ознаки: світле волосся, блакитні очі, шкіра без пігменту, "мишачий" запах (внаслідок виділення із сечею фенілаланіну); після 6 місяців-сонливість, кволість, втрата всіх набутих психомоторних функцій, судоми, формування розумової відсталості аж до ідіотії.
Діагноз ставиться на основі клінічного обстеження та результатів біохімічного виявлення фенілпірувату в сечі і фенілаланіну в крові. Лікування -дієтотерапія: виключення з раціону продуктів, що містять фенілаланін (яйця, м'ясо, молоко). Рання діагностика і дієтотерапія попереджають розвиток клінічної картини хвороби.
Альбінізм - спадкова хвороба - ферментопатія, зумовлена недостатністю фермента тирозинази, що каталізує реакції, необхідні для утворення чорних пігментів - меланінів. Відсутність меланінів у меланоцитах шкіри проявляється недостатньою (або відсутньою) пігментацією шкіри, волосся, підвищеною чутливістю шкіри до сонячного світла, порушенням зору. Тип успадкування — автосомно-рецесивний.
Алкаптонурія - спадкова хвороба автосомно-рецесивного типу успадкування, яка спричинюється генетично детермінованою недостатністю фермента оксидази гомогентизинової кислоти. Характерним проявом захворювання є надмірне виділення гомогентизинової кислоти з сечею, яка при додаванні лугів набуває темного забарвлення. Гомогентизинова кислота нагромаджується в сполучній тканині. Суглобові хрящі набувають жовто- оранжового кольору (охроноз), хрящі вушних раковин і носа темніють, розвиваються артрити.
Тирозиноз (тирозинемії) - дефект тирозин-амінотрансферази або оксидази п- гідроксипіровино-градної кислоти. Супроводжується накопиченням в крові та виділенням з сечею тирозина. Порушення гомеостазу. В гострій формі захворювання характеризується затримкою розвитку малюка, появою гепатоспленомегалії, геморагії, змінами в нирках. Без лікування діти гинуть в грудному віці від печінкової або дихальної недостатності. Для хронічного перебігу хвороби притаманні цироз печінки, рахітоподібні зміни кісток, ураження канальцевої системи нирок. Тип успадкування — аутосомно-рецесивний.
Лейциноз - мутації трьох різних ферментів (декарбоксилази, трансацилази, флавінового ферменту), порушується окислювальне декарбоксилювання 3-х кетокислот, в які перетворюється лейцин, ізолейцин і валін. Клінічно розрізняють декілька форм цієї хвороби — класичну, проміжну, м’яку, тіамінзалежну. Основні симптоми пов’язані з ураженням нервової системи: судоми, порушення дихання, в сечі надлишок кетокислот надає їй запаху кленового сиропу.
До спадкових хвороб - порушень обміну вуглеводів - відносять галактоземію, мукополісахаридози.
Галактоземія — порушення обміну галактози, яка надходить з їжею та утворюється при гідролізі лактози. Тип успадкування А - Р, у гомозигот активність ферменту 3-12 % від норми, у гетерозигот — 50 %. Частота 1:35...150 тис. народжень. Галактоземія характеризується гетерогенністю (різні варіанти мутацій гену). Наприклад, з частотою 1:100...200 тис. зустрічається галактоземія з м’якою клінічною картиною. Фенотипово (клінічно) проявляється жовтяниця новонароджених, блювання, пронос, розвиток розумової відсталості, враження печінки, дистрофія. При ранній діагностиці дитині призначають спеціальну (як і при фенілкетонурії) дієту - виключення молока матері та інших продуктів, які містять лактозу або галактозу. Розвиток нормалізується.
Мукополісахаридози — спадкові хвороби. Порушення метаболізму глікозамінгліканів (ГАГ), накопичення ГАГ внаслідок мутації ферментів лізосом (гідролаз). Генетична гетерогенність визначається мутаціями різних генів, які кодують різні ферменти. Тип успадкування А-Р, Х-Р. Фенотипово (клінічно) проявляються в порушенні розвитку (карликовість, вади обличчя, малорухомість суглобів, зменшення мозку, рання смертність 12-20 років). З сечею виділяється багато мукополісахаридів. Найчастіше зустрічається синдром Гурлера (гаргоілізм), синдром Хантера (мукополісахаридоз, тип ІІ).
Спадкові дефекти обміну ліпідів — сфинголіпідози — порушення розщеплення ліпідів та порушення обміну ліпідів плазми крові. Тип успадкування А-Р, Х-Р. Частота різних форм від ≈ 1:4000 новонароджених до 1:300 000, частота в різних популяціях може значно відрізнятися. Спадкові хвороби пуринів і піримідинів. Приклад синдром Леша-Найяна. Частота 1 : 300000. Тип успадкування може бути Х-Р, А-Р. Порушення обміну — нестача ферменту, необхідного для синтезу ДНК. В сечі хворих накопичується сечова кислота. Фенотипові порушення: розумова відсталість, симпатичні паралічі, порушення пуринового обміну, агресивна поведінка, сечокам’яна хвороба.
Спадкові дефекти обміну вітамінів — гомоцистонурія - генетичний дефект коферменту вітамінів В6 і В12 (піридоксинзалежні ензимопатії). А - Р тип успадкування. Частота в популяціях - 1:50000. Характерна фенотипова різноманітність, яка визначається гетерогенністю. Спостерігаються ураження очей (ектопія кришталика), зміни скелету, розумова відсталість, розширення кровоносних судин, тромбози, порушення функцій ЦНС, недоумкуватість.
Спадкові хвороби — порушення біосинтезу стероїдних гормонів зустрічаються з частотою 1:5000...10000; А-Р тип успадкування. До цієї групи відносять:
адреногенітальний синдром (мутації генів, які контролюють синтез андрогенів -- чоловічих гормонів; тестикулярна фемінізація, при якій не утворюються рецептори андрогенів). При даних хворобах порушується процес диференціації статі (псевдогермафродитизм), аномалії та вади розвитку статевих органів (гіпоспадія, гіпоплазія.).
Гемоглобінопатії — група спадкових хвороб, при яких порушуються білкові ланцюги гемоглобіну (НЬ), що призводить до змін їх функцій і властивостей. До таких хвороб відносяться: метгемоглобінемія, еритроцитози, серпоподібноклітинна анемія, таласемія.
Найвідоміша хвороба серпоподібноклітинна анемія, яка з високою частотою зустрічається в регіонах розповсюдження малярії. Тип успадкування — аутосомний, неповністю домінантний. Мутантний ген (S) викликає синтез гемоглобіну S, який змінює форму еритроцитів (рис) та слабо приєднує кисень, внаслідок чого розвивається анемія та гіпоксія. У гетерозигот — одночасно є нормальний HbS та мутантний HbS, але вони не хворіють на малярію.
Таласемії — хвороби, при яких зменшується вміст білку - глобіну в молекулі гемоглобіну (Hb). Тип успадкування А - Р або внаслідок делецій. Для діагностики виду таласемій використовують молекулярно - генетичний метод, електрофорез.
Колагенові хвороби - в основі цих хвороб є генетичні дефекти біосинтезу та розпаду колагену (структурний компонент сполучної тканини). До цієї групи відноситься хвороба Елерса - Дамлоса, для якої характерний генетичний поліморфізм, тип успадкування А-Д, А-Р; хвороба Марфана (А-Д тип успадкування). Фенотипово плейотропна дія мутантних генів проявляється гіпермобільним синдромом, збільшеною еластичністю шкіри, внутрішніми кровотечами, змінами в суглобах, блакитними склерами. Первинні дефекти -порушення біосинтезу колагену або процесингу фібрил і колагену.
До спадкових генних хвороб з невідомим первинним біохімічним дефектом відносять:
1) Ахондроплазію (А-Д тип успадкування, частота 1:100 000; виникає внаслідок мутації de novo). Фенотипово проявляється порушеннями скелету (порушення утворення хрящової тканини в епіфізах трубчастих кісток, кісток черепу).
2) Муковісцидози (А-Д або А-Р тип успадкування, частота 1:2500 новонароджених). В основі патогенезу всіх форм — ураження ендокринних залоз (секретуючих клітин бронхів, підшлункової залози, кишечнику, потових залоз, печінки), що супроводжується виділенням густого секрету, запальними та склеротичними змінами в органах. Основні форми — легенева та кишкова. Діагностика — спеціальні комплексні тести (визначення вмісту Na в секретах, визначення активності травних ферментів...). Вважається, що значна кількість випадків у дітей не діагностується.
3) Міопатії (м’язові дистрофії) — група спадкових хвороб, при яких уражаються посмуговані та гладенькі м’язи. Тип успадкування може бути Х-Р, А-Д, А-Р. Для міопатій характерне ураження м’язів, яке з віком прогресує, клінічний поліморфізм.
Форми еритроцитів людини

Рис. 35. Зміни форми еритроцитів при серпоподібноклітинної анемії.
Хвороби зі спадковою схильністю. Хвороби зі спадковою схильністю (мультифакторіальні хвороби) зумовлені комбінацією генетичних і негенетичних факторів. Останні пов'язані із зовнішнім середовищем. Для реалізації цих хвороб необхідна не лише відповідна генетична конституція індивідуума, але і фактор або комплекс факторів середовища, які відіграють роль пускових моментів у формуванні патології. До таких захворювань відносяться: атеросклероз, подагра, ревматизм, ішемічна хвороба серця, гіпертонічна хвороба, епілепсія, виразкова хвороба шлунка і 12-палої кишки, цироз печінки, цукровий діабет, бронхіальна астма, туберкульоз, псоріаз, шизофренія, ін.
Характерні ознаки мультифакторіальних хвороб: 1) великий поліморфізм клінічних форм та індивідуальних проявів; існування перехідних форм від здорових людей до хворих, від субклінічних форм до тяжкого перебігу; 2) висока частота в популяції (на цукровий діабет страждає 5 % людей земної кулі, на алергічні захворювання - понад 10 %, на шизофренію - 1 %, на гіпертонію - близько 30 %); 3) невідповідність успадкування законам Г. Менделя; 4) різний вік хворих.
По спадковості передається схильність до певного захворювання. Для деяких клінічних форм роль спадкового (сімейного) фактору є вирішальною. Ступінь ризику для родичів хворого залежить від частоти хвороби в популяції і зростає з тяжкістю перебігу в нього. Чим ближче ступінь спорідненості з хворим у родичів, тим більша ймовірність народження в них хворої дитини. У ряді випадків спостерігається неоднакова частота патології залежно від статі. Наприклад, уроджена дисплазія кульшового суглоба частіше зустрічається в дівчаток, а пілоростеноз - у хлопчиків.
Хвороби зі спадковою схильністю можуть бути моногенними і полігенними. Основу складає полігенне успадкування і часто гетерозиготність. При полігенному успадкуванні ознаку зумовлюють кілька неалельних генів, але прояв їх залежить від умов середовища. При гетерозиготному носійстві патологічний рецесивний ген у гетерозиготному стані не проявляється, але може проявитися за несприятливих умов життя. Оскільки хвороби зі спадковою схильністю визначаються поєднанням спадкових і зовнішніх факторів, їх відносять до захворювань з пенетрантністю, яка в значній мірі залежна від умов середовища. Змінюючи умови середовища, можна значно змінити прояв таких хвороб і навіть попередити їх. Моногенні хвороби багаточисельні (>3000 різних хвороб), характеризуються гетерогеністю (різними мутаціями будь-яких ділянок гену відповідного ферменту, або різних неалельних генів) та фенотиповий (клінічним) поліморфізмом. Гетерогенність встановлена для більшості моногенних хвороб, наприклад, для фенілкетонурії (>15 форм), галактоземії (>12 форм), альбінізму (>6 форм) та інших моногенних хвороб (на фенотиповому рівні гетерогенність проявляється різними варіантами клінічних проявів спадкової патології). Для деяких хвороб гетерогенність визначається наявністю генокопій (мутацій різних неалельних генів, які визначають однакові порушення обміну речовин), що також визначає генетичну різноманітність, клінічний поліморфізм спадкових хвороб. Приклади генокопій у людини: фенілкетонурія класична та дієторезистентна форми; різні форми гемофілій, колагенозів.
Біохімічні методи
Біохімічні методи дозволяють діагностувати спадкові хвороби, обумовлені генними мутаціями, які викликають порушення обміну речовин або їх структури, а також поліморфізм у молекулярних хвороб.
Існує два напрямки:
1) вивчення продуктів обміну генетично детермінованих процесів (збільшення або зменшення концентрації певних продуктів обміну вказує на зміну активності ферменту, його мутації);
2) встановлення порушень структури транспортних білків, ферментів.
Біохімічну діагностику спадкових порушень проводять в 2 етапи. На першому (просіючому) етапі відбирають експрес методами найбільш ймовірні випадки хвороб, на другому — більш складними методами уточнюють діагноз. Для експрес діагностики зазвичай використовують мікробіологічне тестування.
Біохімічні методи використовують також в пренатальній або післяпологовій діагностиці моногенних хвороб, що дозволяє своєчасно виявити патологію та призначити специфічні медичні заходи (профілактичні або лікувальні).
Генна інженерія
Генна (генетична) інженерія - сукупність експериментальних методів, за допомогою яких переносять гени з одного організму в інший з метою спрямованого надання останньому нових спадкових ознак. Для генетичної інженерії не існує таксономічних бар'єрів. Вона дозволяє маніпулювати з генетичним матеріалом з різних джерел і за заданою програмою конструювати in vitroфункціонально активні рекомбінантні (гібридні, хімерні) молекули ДНК, які не зустрічаються в природі. Префікс "ре" означає, що ДНК не створюється de novo (заново), а утворюється внаслідок об'єднання фрагментів вже існуючих молекул. Хімерними рекомбінантні молекули ДНК називаються тому, що можуть поєднувати, здавалось б, несумісні гени від різних організмів. Теоретичну основу генетичної інженерії складає універсальність генетичного коду.
Генетична інженерія включає наступні етапи: 1) одержання генів шляхом штучного (хімічного або матричного) синтезу або шляхом виділення їх з природних джерел; 2) включення гена у векторну молекулу ДНК, тобто створення рекомбінантних молекул ДНК; 3) введення векторної молекули ДНК з включеним геном у реципієнтну клітину; 4) створення умов для експресії перенесеного гена та його стабільного успадкування; 5) відбір клітин з діючим перенесеним геном - молекулярне клонування.
Штучний синтез гена хімічним шляхом вперше здійснив у 1969 р. індійський учений Г. Корана з співробітниками. Це був ген аланінової тРНК дріжджів з 77 пар нуклеотидів. Але синтезований ген не містив регуляторної частини і тому був функціонально не активним. Пізніше ці автори синтезували функціонально активний ген - ген супресорної тирозинової тРНК Е.соlі довжиною близько 200 пар нуклеотидів. Хімічному синтезу генів сприяла розробка методів встановлення первинної структури ДНК, тобто послідовності нуклеотидів в її молекулі (секвенування). Метод хімічного синтезу гена відкрив широкі можливості для штучного синтезу генів людини. Ген гормону росту людини (соматотропін), ген інсуліну людини одержані хімічним синтезом.
Штучний синтез гена матричним шляхом здійснюється за допомогою фермента зворотної транскриптази (ревертази). Фермент здатний будувати копії ДНК на різних РНК, включаючи синтетичні. Матричним шляхом можна синтезувати практично будь-який ген на матриці іРНК. Таким шляхом синтезований ген інтерферона людини - цінного лікарського препарата, який застосовується для боротьби з вірусними інфекціями.
Метод виділення гена з природної ДНК ґрунтується на інкубації тотальної ДНК з різними рестрикційними ендонуклеазами (рестриктазами). Рестриктази - це ферменти ("молекулярні ножиці"), які розрізають молекулу ДНК у специфічних сайтах на фрагменти (рестрикція). Фрагменти розділяють методом електрофорезу, виділяють у чистому вигляді і визначають послідовність нуклеотидів.
Після одержання генів шляхом синтезу або виділення з природних джерел наступним етапом генної інженерії є включення необхідного гена у векторну молекулу ДНК. Роль векторів виконують плазміди, бактеріофаги, деякі віруси, мітохондріальна ДНК. Найбільш часто в якості векторів застосовують плазміди. Плазміди - позахромосомні невеликі молекули ДНК кільцевої форми, здатні до автономної реплікації; вони розташовані в цитоплазмі бактеріальної клітини або інтегровані в її хромосому і тоді називаються епісомами. Епісоми відтворюються в складі хромосоми. Векторну молекулу ДНК кільцевої форми розрізають рестриктазами на лінійні фрагменти. Фрагменти векторної ДНК і фрагменти чужорідної ДНК своїми комплементарними ("липкими") кінцями здатні об'єднуватися в одну рекомбінантну (гібридну) молекулу ДНК. Фосфодіефірні зв'язки між нуклеотидами утворюються за допомогою ферментів лігаз. Перенесення необхідних генів (трансгеноз) забезпечується різними способами: трансформацією (якщо вектор - плазміда), трансдукцією (вектор - бактеріофаг).
Методами генної інженерії одержані трансгенні рослини і тварини (організми з чужорідними генами). Трансгенні тварини використовуються в біомедицині як моделі захворювань людини (миші з геном раку, свині з людськими патологіями серця, корови з людськими имуноглобулінами в крові). Розв'язується проблема одержання людських білків крові в молоці трансгенних тварин.
Своїми успіхами генна інженерія в значній мірі зобов'язана створенню банків (бібліотек) генів. Банком генів називають набір генів, одержаний на основі рекомбінантних молекул. Генетик може відібрати в бібліотеці генів необхідні для дослідження гени за допомогою спеціально розроблених генетичних, біохімічних, радіоізотопних чи імунологічних методів. Створені банки генів дрозофіли, кишкової палички і багатьох інших організмів, у тому числі і людини.
Генетична інженерія народилася в 1972 p., коли американські генетики П. Берг, Г. Бойер і С. Коен створили in vitro першу рекомбінантну ДНК, яка об'єднала в своєму складі генетичний матеріал з трьох різних джерел: повний геном онкогенного вірусу мавпи SV40, частину генома помірного бактеріофага X (лямда) і гени галактозного оперона кишкової палички (Е.соlі). Проте сконструйована рекомбінантна молекула не була досліджена на функціональну активність, оскільки в авторів виникли побоювання, що методи генної інженерії можуть привести до створення організмів, небезпечних для здоров'я людини. Втручання в генотип організму може привести до непередбачених наслідків для людини, рослин, тварин і довкілля в цілому. Наприклад, бактерія кишкова паличка, нешкідлива в звичайних умовах, може перенести онкогенні віруси тварин у кишечник людини. Спеціально сконструйовані біологічні агенти, які уражають живе, розглядаються як біологічна зброя. На Міжнародній конференції з біобезпеки в м. Асиломарі (США) в 1975 р. були розроблені правила, дотримання яких усуває ймовірність шкідливих наслідків генної інженерії. У 1985 р. сформована Інформаційна робоча група з біобезпеки.
Біотехнологія
Біотехнологія (грец. bios - життя, techne - майстер, logos - наука) - наука, яка вивчає застосування живих організмів або біологічних процесів у промисловості. Біотехнологічні методи відомі людству давно. У таких галузях промисловості як виноробство, хлібопекарство, броварство (пивоваріння), сироваріння широко використовуються мікроорганізми. З метою одержання біологічно активних речовин (антибіотиків, гормонів, ферментів, вакцин), сучасна біотехнологія ґрунтується на застосуванні останніх досягнень генетичної інженерії в галузі створення рекомбінантних молекул ДНК. У фармацевтичній промисловості виникла нова галузь - індустрія ДНК. У мікробіологічній промисловості використовують трансгенні штами бактерії кишкової палички (Е.соlі), у геном яких методами генетичної інженерії введені гени людини, які кодують синтез інсуліну, інтерферону, соматотропіну людини. І бактерії в промислових умовах синтезують ці лікарські речовини. Мікробіологічний синтез - надзвичайно ефективний процес. Так, щоб одержати 5 мг соматотропіну (гормон росту), потрібно використати мозок 500 000 овець протягом 5 років, у той час як аналогічну кількість гормону дають 9 л бульойнної суспензії кишкової палички.
Перспективи гемотерапії
Генна терапія - це метод введення фрагмента ДНК у клітини хворої людини з метою заміщення функції мутаційного гена і лікування спадкових хвороб. Ще в кінці 60-х років виявили, що клітини тварин і людини здатні поглинати екзогенну ДНК, вбудовувати її в свій геном, після чого проявляється експресія введених генів, зокрема у вигляді синтезу відсутніх раніше білків і ферментів. Були розроблені методи доставки ДНК у клітини за допомогою вірусів та інших носіїв.
Перша спроба генної терапії в клініці була здійснена М. Клайном у 1983 p., коли він ввів нормальний р-глобіновий ген хворим р-таласемією. Пізніше була розроблена методика генної терапії спадкової недостатності аденозиндезамінази (тяжкий імунодефіцит): нормальний ген був введений у клітини кісткового мозку хворого і після їх ретрансплантації відновилась активність ферменту, так що стан хворого поліпшився. Проведені клінічні експерименти з генотерапії раку. У лейкоцити хворих на злоякісну меланому і пізні стадії раку були введені гени, які мітили (маркували) злоякісні клітини, щоб їх могла розпізнати імунна система. У половини хворих розміри пухлин зменшились у два рази і більше. Для 40 хвороб розроблені методи генної терапії. У найближчий час цей метод буде поширюватись, оскільки розшифрований геном людини.